内部陽性対照
はじめに
リアルタイムPCRを使用してマルチプレックスリアルタイムPCRのためのFRET-TaqManプローブの設計は、閉じた管形式でPCR産物の生成中にフルオロフォア蛍光の継続的なモニタリングを可能にする。 現在、利用可能な方法は、PCR産物の増幅をモニターするために標識されたプローブまたはDNAインターカレート色素のいずれかを利用する。 TaqManプローブは、フルオロフォアとクエンチャーを含む一本鎖オリゴヌクレオチドであり、10-30塩基離れて配置されている。 TaqManプローブは、蛍光増加の結果、クエンチャーからフルオロフォアを分離Taqポリメラーゼの5′-3’二本鎖特異的エキソヌクレアーゼ活性のために増幅中に加水分解 リアルタイムPCR機器には、蛍光検出器が装備されており、陽性反応のために、蛍光が背景蛍光よりも大きいサイクルであるサイクル閾値(Ct)を推定する しかしながら、この技術は、PCRが増幅阻害剤によって影響される場合には、真の陰性結果を偽陰性と区別することができない。 核酸抽出後、阻害剤は、臨床試料(例えば、ヘモグロビン)、環境試料(例えば、フミン酸およびフルボ酸)、および核酸抽出中に使用される化学物質(例えば、エタノール、洗剤、またはカオトロピック剤)から依然として存在し得ることが報告されている。 診断アッセイの信頼性は、PCR阻害剤の存在および影響を示すことができる内部対照核酸を含めることによって増加する(1,2)。 内部陽性対照(IPC)は、ターゲット配列アッセイに使用されるフルオロフォアとは異なる波長で光を放射する標識されたフルオロフォアを使用して、標的配列の存在下で同時に増幅され、リアルタイムPCR装置によって異なるチャネルで検出された二つのフルオロフォアを使用する。 PCRのために一般的に使用される内部対照は、プローブ領域を除いてアッセイ標的の配列と同様の配列を含むプラスミドである。 限られた数のIPC分子が個々のアッセイ標的に添加され、標的核酸と共増幅される。; 従って、正のIPCシグナルは、増幅反応が非常に少量の標的核酸から正のシグナルを生成するのに十分に進行した証拠である。 この特徴は、IPCおよび標的核酸の同等の増幅を確実にするために重要である(3−9)。 2およびLightcycler2(Roche Applied Science,Indianapolis,IN,USA)、Ruggedized Advanced Pathogen Identification Device(R.)器械(アイダホの技術、ソルトレイクシティ、UT、米国)、および手持ち型の実時間PCRの器械は-一つの光源および準の放出チャネルがだけ装備されています。 これらのタイプの器械の多重型になることは複数の分類されたオリゴヌクレオチドおよび蛍光性共鳴エネルギー移動(FRET)の概念の使用を要求する。 しかし、FRETプローブは設計が困難であり、超可変領域および最適でない増幅条件が存在する場合にはロバスト性を欠く可能性がある。 FRETプローブを使用するには、2つのプローブに対して2つの領域を識別する必要があります。 FRETプローブシステムは,アクセプター色素の蛍光が第二プローブに付着したフルオロフォアの励起によって検出されるような方法で,二つの単一標識プローブのオリゴヌクレオチドの隣接ハイブリダイゼーションに依存している。 FRETプローブの主な欠点は、2-5ヌクレオチドのギャップを持つ二つの隣接するプローブを収容するために必要なより大きな配列領域の要件です。 Megastokes dye(DYXL;Dyomics,Jena,Germany)およびPulsar6 5 0(Biosearch Technologies,Inc.,Novato,CA,USA)が、これらのラベリングシステムは、チャネルのクロストークを補償するためにスペクトル情報を必要とし、広く利用可能ではありません。 しかし、本研究で開発されたFRET-TaqManプローブは、色補正ソフトウェアを必要とせず、広く利用可能なfluorophores(FAM、Cy5.5)を使用しています。 本報告書は、唯一の青色発光ダイオード(LED)励起源(470nm)と関連する三つの対応する検出チャネル(例えば、530nm、640nm、および705nm)を備えた機器で多重化を可能にするために三重標識プローブの設計に焦点を当てている。
材料と方法
プローブ設計原理
すべての実験は、アイダホテクノロジのR.A.P.I.D.システム、励起用の青色LED(470nm)とモノカラーアッセイ用の530、640、および705nmで蛍光を測定する関連する三つの蛍光検出チャネルを備えた32サンプルのポータブルリアルタイムPCR装置で行われた。 プライマー JVAFおよびJVAR(1 0)ならびにプローブ5’−FAM/BHQ−1を、CDC Biotechnology Core Facility(Atlanta,G A,USA)で合成し、プローブ5’FAM−Cy5. 二重鎖Taqman PCRを可能にするために、1つのプローブを5’−FAM/BHQ−1(5 2 0nmの蛍光波長)で標識し、別のプローブを5’−Fam−Cy5.5/3’BHQ−3(7 0 5nmの蛍光波長)で標識した。 溶液中で、5’FAM-Cy5.5/3’BHQ-3で標識されたプローブは、配列へのハイブリダイゼーション時にクエンチされたままであり、酵素プライマー伸長によるプローブのその後の切断時に、クエンチャーは排除される。 これはR.A.P.I.D.の器械の光源によってFAMの刺激によって焦燥で起因する。 励起エネルギーは、アクセプタ蛍光体Cy5に伝達される。図5に示すように、放出された蛍光は、第二の蛍光チャネル(R.A.P.I.D.機器の蛍光チャネルF3)で測定される。 Cy5.5蛍光の放出は、FRETおよびTaqmanプローブ技術の組み合わせの結果であるので、これらの三重標識プローブは、「FRET−Taqman」プローブと呼ぶことができる。
プライマー、プローブ、およびプラスミド
フレット-TaqManデザインの多重化能力を実証するために、我々は18S rRNA遺伝子(159bpの部分を増幅するために属特 フォワードプライマー(JVAF)は5’−ATGACGGGTAACGGGGAAT−3’であり、リバースプライマー(JVAR)は5’−CCAATTACAAAAC−CAAAAAAGTCC−3’であり、Taqmanプローブ(JVAP)は5’−FAM−CGCGCCTGCT−GCCTTCCTTAGATG−BHQ1−3’であり、ヌクレオチド1 0 0−1 1 8、2 5 8−2 3 6および1 8 4−1 6 1に対応した。それぞれ、genbank ACCESSION no. 458612 Cryptosporidium IPCの構築のためのプライマーは,Cryptosporidium sppの増幅のために報告されたプライマー配列と同じセットを含んでいた。 標的DNAのプローブ結合領域を除いて、人工配列5’−TTGTGCTCGTAGTCGCCTCTGTCTCT−3’(JPIPC)に置換した。 このプローブシーケンスは69℃のアニーリング温度を有し、二次構造を持たないように設計された。 内部プローブの理論的特異性は、完全に一致する任意のCryptosporidium配列と発見されなかったプローブとプライマー配列のBLAST(11)検索によって確認されました。 IPC断片を合成し、Integrated DNA Technologies(Coralville,I A,USA)によってプラスミドにクローニングした。 既知の濃度のプラスミドDNA(1 0 0コピー/反応)を、陰性対照を除く全ての反応のマスター混合物に添加した。 IPCに特異的な三重標識プローブ(JPIPC)を、図1に示すように、Idaho Technologyによって合成した。 FAM/Cy5.5/BHQ3オリゴヌクレオチドプローブは、非蛍光ブラックホールクエンチャー(BHQ-3)制御細孔ガラス(CPG)と3’末端に開始して合成されました。 次いで、5’末端をCy5.5ホスホラミダイトと結合させ、続いて末端5’フルオレセイン(6-FAM)ホスホラミダイトと結合させた。
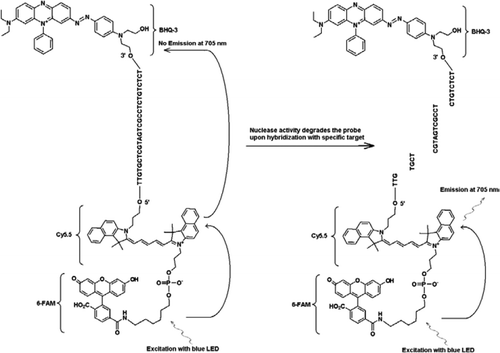
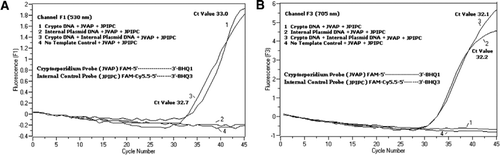
FRET-TaqManプローブには、3’アームの端にブラックホールクエンチャー部分、エミッタフルオロフォア(Cy5.5)、および5’アームの端に結合したハーベスタフルオロフォア(FAM)の三つのラベルが含まれている。 FAMは効率的に光源として青いLEDからのエネルギーを吸収する。 ターゲットがない場合、famによって吸収されたエネルギーがCy5.5に伝達され、次にcy5から放出されるエネルギーが伝達されるため、プローブは暗くなります。5はクエンチャーに移され、熱エネルギーとして失われます。 ターゲットの存在下では、TaqManプローブはTaqポリメラーゼの活性によって切断され、クエンチャーが放出され、FAMによって吸収されたエネルギーはFRET機構を介してCy5.5に転送され、705nmで蛍光を放出する。University o f Wisconsin’s State Laboratory o f Hignity(Madison,WI,USA)から入手したCryptosporidium parvumオーシスト(チューブあたり2 0 0オーシスト)を選別したフローサイトメトリー。 CからのDNA。 parvumオーシストをHillらによって記載されたプロトコールを用いて抽出した。 (1 2)Tris EDTA(TE、pH8. これは5つのオーシスト(標的DNAの100コピーに相当)に対応した。 増幅反応混合物は、Quantifast Probe PCR+ROX kit反応混合物(Cat. roxを添加しないQiagen)、JVAF、JVARプライマー(各0.4μ M)、JVAPプローブ(0.2μ M)、JPIPCプローブ(0.2μ M)、およびIPCの1 0 0コピー。 抽出されたDNAサンプルのアリコート(2μ l)を、1 8μ lの反応混合物を含む反応キャピラリー(Roche Diagnostics,Indianapolis,IN,USA)に加えた。 C.parvumゲノムDNA陽性対照(図2、プロット1)、プラスミドIPC(図2、プロット2)、C.parvum陽性対照とIPCの混合物(図2、プロット3)、およびヌクレアーゼフリー水からなるno-template対照(NTC)(Cat. いいえ。. DNAの代わりにA M9 9 3 2;Ambion,Foster City,C A,USA)(図2、プロット4)を各実験に含めた。 プロトコルは、以下のPCR条件で完了するのに30分以上かかりました: 95°cで3分間のホットスタート変性ステップ、続いて45サイクルで95°c変性を5秒間、60°Cアニーリングを30秒間、単一蛍光獲得(温度ランプレート20°C/s)で行う。 陽性の結果は、チャネルF1(5 3 0nm)でFAMについて、およびチャネルF3(7 0 5nm)でCy5. 増幅反応は三重に行った。 アンプリコン(1 5 9bp)を、アガロースゲル電気泳動およびGlow−Green核酸染色(Eenzyme,Gaithersburg,MD,USA)によって可視化して、PCR産物の特異性を確認した。
結果と考察
TaqManプローブのFAMラベリングにより、C.parvumゲノムDNAの増幅を反映するリアルタイムPCR蛍光シグナルがF1チャネル(図2A)で検出された。 IPCの増幅を反映する蛍光信号は、FAM/Cy5によるF3チャネル(図2B)で検出された。IPC Taqmanプローブの標識化。 このプローブは、エネルギーがFamからCy5.5に転送されたため、BHQ3がヌクレアーゼ活性のためにFAM/Cy5.5から分離されたときにF3チャネル(Cy5.5)でCy5.5の増加が、f1チャネル(FAM)でfamの増加を生じなかった。 同様に、二重反応では、C.parvumゲノムDNA増幅はF1チャネルで検出され、IPC増幅はF3チャネルで同時に検出された。 FAMは、4 9 5nmでの励起および5 2 0nmでの発光を有する。 Cy5.5は690nmの励起と705nmの発光を持っています。 三重標識されたIPCプローブ上のCy5.5とFAMの間にこのような近接性があると、Cy5.5フルオロフォアはFAMフルオロフォアからのエネルギーの完全な移動に 陰性対照反応は、FAMまたはCy5.5による蛍光の増加を示さなかった。 これらの結果はまた単一の青(470nm)LEDの源がFAMおよびCy5.5のためのそれぞれの監視チャネルの分光重複なしでF1(530nm)およびF3(705nm)チャネルを通 FRET−Taqman技術はまた、糞便標本からの1 2個のDNA抽出物のパネル(以前にCryptosporidium sppに対して陽性であると決定された標本からの8個)を用いて試験した。 Cryptosporidiumspp陰性であった四つの標本を示した。). 18S rRNAアッセイは、すべてのエイトCryptosporidium sppを検出することができました。 IPCTAQMANアッセイはIPCプラスミドDNAのみを増幅することが分かった。 IPCアッセイは、3 2〜3 7のCt値をもたらし、これは、いくつかの糞便試料についてのPCR阻害の最大5のCt値を示した(3 2は、非阻害反応について予想されるCt値
本技術は、一方のプローブがドナーのフルオロフォアを含み、他方のプローブがアクセプターのフルオロフォア(13,14)を含む二つのプローブ(FRETプローブ)の代わりに、一方の蛍光標識されたオリゴヌクレオチドプローブ(FRET-TaqManプローブ)が使用される点で、伝統的に使用されているFRETとは異なる。 FRET-TaqManプローブは、5’末端にドナー(FAM)とアクセプター(Cy5.5)フルオロフォアの二つのフルオロフォアで標識され、3’末端にTaqManプローブのようなBHQ-3クエンチャーで標識されている。 ユニークなプローブ結合領域は、色補正ソフトウェアを含める必要なく、同じ増幅反応から二つの異なる検出チャネル(F1で530nmとF3で705nm)における二つの異 標的DNAとIPCは同じプライマー結合部位を持っていたが,プローブ配列は異なっていた。 これにより、プライマー-二量体形成またはプライマーセット間の相互作用を最小限に抑えるために、両方のDNA標的に同じプライマーの使用が保証される。 FAM/Cy5.5フルオロフォアが5’末端に密接に配置されている場合、共鳴エネルギー移動は、Cy5.5フルオロフォアの蛍光発光の増加に伴って行われます。 しかし、BHQ3、Cy5.5のための非蛍光アクセプターの使用は、より高い信号対雑音比をもたらす潜在的な背景蛍光を減少させる。 これは、ゲノムCの≥100コピーを含む反応にIPCの100コピーの添加ことが判明しました。 parvum DNAは、Cryptosporidium TaqManアッセイがシングルプレックスアッセイとして、ipc TaqManアッセイとの二重アッセイとして行われたときに同様のCt値(Ct=32)をもたらした。 Cryptosporidium陽性DNA標本のパネルに適用すると、IPCは、いくつかの標本がPCR阻害剤を含まないことを示した(32の予想Ct値に基づいて)が、他の標本は5Ct値までの阻害
新しく開発された内部PCR陽性コントロールは、R.A.P.I.D機器のF1およびF3蛍光チャネルを監視するためのデュアルカラー多重PCRにおける各IPCおよび18S rRNA標的遺伝子のFAM/Cy5.5-BHQ3およびFAM/BHQ1で標識することにより、同じ反応における二つの標的の同時検出に敏感で特異的であることが判明した。 5’FAM-Cy5.5/3’BHQ-3標識プローブを第二のレポーターとして使用することは、二つのプローブの放出最大値が互いに十分に間隔をあけられていたので有利である。 FRET-TaqManトリプル標識プローブ設計は、一方の青色LED励起と530nmと705nmの二つの発光源からなる光学系を備えた安価で使い捨ての”lab-on-a-chip”デバイスの開発に適用することができ、一方のチャネルでFAMを検出し、他方のチャネルでCy5.5を検出するための適切な光学フィルタを備えている。 この組み合わせは、2つの強力な蛍光源と検出システムを必要としません。
謝辞
著者らは、DNA Sequencing and Genomic Core Facility(ユタ大学、ソルトレイクシティ、UT、米国)のMike PowersとIdaho Technology,IncのRachelle Mullerの支援を認めています。 R.A.P.I.Dサイクラーのための内部肯定的な制御の開発の議論のため。 この出版物は、疾病管理予防センター、テロ準備と緊急対応のための調整オフィスを通じて利用可能な資金によって部分的にサポートされていました。 商標名および商業ソースの使用は、識別のみを目的としており、疾病管理予防センターまたは米国保健福祉省による承認を意味するものではありません。
- 1. Hartman、L.J.、S.R.Coyne、およびD.A.Norwood。 2005. Taqmanベースのアッセイのための新規な内部陽性対照の開発。 モル セル 19:51-59Crossref,Medline,CAS,Google Scholar
- 2. Hoorfar、J.、B.Malorny、A.Abdulmawjood、N.Cook、M.Wagner、およびP.Fach。 2004. 診断PCRアッセイのための内部増幅制御の設計における実用的な考慮事項。 J.Clin. ミクロビオール 42:1863–1868.Crossref,Medline,CAS,Google Scholar
- 3. Betsou,F.,K.Beaumont,J.M.Sueur,j.Orfila. 2003. 尿サンプルからのクラミジアtrachomatis DNAのPCR増幅のための内部制御DNAの構築と評価。 J.Clin. ミクロビオール 41:1274–1276.Crossref,Medline,CAS,Google Scholar
- 4. Burggraf,S.およびB.Olgemoller. 2004. 実時間拡大の試金の内部制御のための簡単な技術。 クリン ケム 50:819–825.Crossref,Medline,CAS,Google Scholar
- 5. Drosten,C.,M.Weber,E.Seifried,And W.K.Roth. 2000. 献血者のスクリーニングのための競争の内部制御順序との新しいPCRの試金の評価。 40:718-724Crossref,Medline,CAS,Google Scholar
- 6. グルーバー、F.、F.G.フォークナー、F.ドーナー、T.ハマーレ。 2001. 二重増幅、内部標準化、および二色蛍光検出を適用したリアルタイムPCRによるウイルスDNAの定量化。 Appl. エンビロン ミクロビオール 67:2837–2839.Crossref,Medline,CAS,Google Scholar
- 7. ローゼンストラウス、M.、Z. Wang、S.Y.Chang、D.DeBonville、およびJ.P.Spadoro。 1998. 定期的な診断PCRのための内部制御:臨床性能に対する設計、特性および効果。 J.Clin. ミクロビオール 36:191–197.Medline、CAS、Google Scholar
- 8。 ることができる。 2003. 実時間PCRの試金のパネルのための多数の内部制御DNAの生成への便利なアプローチ。 J.ヴィロール 108件中1-8件を表示しています。Crossref,Medline,CAS,Google Scholar
- 9. およびJ.W.Mannhalter。 1996. 定量的競争力のあるPCRの技術的側面。 BioTechniques21:268-279.リンク、CAS、Google Scholar
- 10。 Jothikumar,N.,A.J.da Silva,I.Moura,Y.Qvarnstrom,V.R.Hill. 2008. 二重TaqManの試金によるCryptosporidiumのhominisおよびCryptosporidiumのparvumの検出そして微分。 J.Med. ミクロビオール 57:1099–1105.Crossref、Medline、CAS、Google Scholar
- 11。 ることができると考えられている。 1990. 基本的なローカル整列検索ツール。 J.モル。 バイオル 215:403–410.Crossref、Medline、CAS、Google Scholar
- 12。 Hill,V.R.,A.M.Kahler,N.Jothikumar,T.B.Johnson,D.Hahn,t.L.Cromeans. 2007. 100リットルの水道水サンプル中の腸内微生物の同時回収のための限外ろ過ベースの手順の多州評価。 Appl. エンビロン ミクロビオール 73:4218–4225.Crossref、Medline、CAS、Google Scholar
- 13。 Bernard,P.S.,M.J.Lay,C.T.Wittwer. 1998. 蛍光共鳴エネルギー移動および調査の溶けるカーブによるmethylenetetrahydrofolateの還元酵素の遺伝子のC677Tポイント突然変異の統合された拡大そして検出。 アナル… バイオケム 255:101–107.Crossref、Medline、CAS、Google Scholar
- 14。 Wittwer,C.T.,M.G.Herrmann,A.A.Moss,And R.P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130–138.Link, CAS, Google Scholar