Design von FRET-TaqMan-Sonden für die Multiplex-Echtzeit-PCR unter Verwendung einer internen Positivkontrolle
Einführung
Die Echtzeit-PCR ermöglicht die kontinuierliche Überwachung der Fluorophorfluoreszenz während der Erzeugung von PCR-Produkten in einem geschlossenen Röhrchenformat. Derzeit verfügbare Methoden verwenden entweder markierte Sonden oder DNA-interkalierenden Farbstoff, um die Amplifikation des PCR-Produkts zu überwachen. Die TaqMan-Sonde ist ein einzelsträngiges Oligonukleotid, das einen Fluorophor und einen Quencher enthält, die im Abstand von 10-30 Basen angeordnet sind. Die TaqMan-Sonde wird während der Amplifikation aufgrund der 5′-3′ doppelstrangspezifischen Exonukleaseaktivität der Taq–Polymerase, die den Fluorophor vom Quencher trennt, hydrolysiert, was zu einer Fluoreszenzzunahme führt. Echtzeit-PCR-Instrumente sind mit Fluoreszenzdetektoren und Software ausgestattet, die in der Lage sind, die Zyklusschwelle (Ct), den Zyklus, bei dem die Fluoreszenz größer ist als die Hintergrundfluoreszenz, für positive Reaktionen abzuschätzen. Die Technik kann jedoch kein wirklich negatives Ergebnis von einem falsch negativen unterscheiden, wenn die PCR durch Amplifikationsinhibitoren beeinflusst wird. Nach der Nukleinsäureextraktion wird berichtet, dass Inhibitoren aus klinischen Proben (z. B. Hämoglobin), Umweltproben (z. B. Humin- und Fulvosäuren) und Chemikalien, die während der Nukleinsäureextraktion verwendet werden (z. B. Ethanol, Detergenzien oder Chaotropika), noch vorhanden sein können. Die Zuverlässigkeit diagnostischer Assays wird durch die Aufnahme einer internen Kontroll-Nukleinsäure erhöht, die das Vorhandensein und die Wirkung von PCR-Inhibitoren anzeigen kann (1,2). Eine interne Positivkontrolle (IPC) wird gleichzeitig in Gegenwart einer Zielsequenz unter Verwendung eines markierten Fluorophors amplifiziert, der Licht mit einer anderen Wellenlänge als der für den Zielsequenztest verwendete Fluorophor emittiert, wobei die beiden Fluorophore in verschiedenen Kanälen durch das Echtzeit-PCR-Instrument nachgewiesen werden. Die häufig verwendete interne Kontrolle für die PCR ist ein Plasmid, das eine Sequenz enthält, die der des Assay-Targets mit Ausnahme der Sondenregion ähnlich ist. Eine begrenzte Anzahl von IPC-Molekülen wird dem einzelnen Assay-Target zugesetzt und mit der Zielnukleinsäure co-amplifiziert; somit ist ein positives IPC-Signal ein Beweis dafür, dass die Amplifikationsreaktion ausreichend verlief, um ein positives Signal aus sehr kleinen Mengen von Zielnukleinsäure zu erzeugen. Dieses Merkmal ist wichtig, um eine äquivalente Amplifikation des IPC und der Zielnukleinsäure (3-9) sicherzustellen. Einige Geräte — wie der LightCycler 1.2 und LightCycler 2 (Roche Applied Science, Indianapolis, IN, USA), das Ruggedized Advanced Pathogen Identification Device (R.A.P.I.D.) Instrument (Idaho Technology, Salt Lake City, UT, USA) und Handheld-Echtzeit—PCR-Instrumente – sind nur mit einer Lichtquelle und einem zugehörigen Emissionskanal ausgestattet. Multiplexing auf diesen Arten von Instrumenten erfordert die Verwendung von mehrfach markierten Oligonukleotiden und das Fluoreszenz-Resonanz-Energie-Transfer (FRET) -Konzept. FRET-Sonden können jedoch schwierig zu entwerfen sein und können in hypervariablen Regionen und bei nicht optimalen Verstärkungsbedingungen keine Robustheit aufweisen. Die Verwendung von Bundsonden erfordert die Identifizierung von zwei Regionen für zwei Sonden. Das FRET-Sondensystem beruht auf benachbarter Hybridisierung von Oligonukleotiden der beiden einzelmarkierten Sonden derart, dass die Fluoreszenz des Akzeptorfarbstoffs durch die Anregung des an der zweiten Sonde angebrachten Fluorophors detektiert wird. Ein wesentlicher Nachteil der FRET-Sonde ist das Erfordernis eines größeren Sequenzbereichs, der notwendig ist, um zwei benachbarte Sonden mit einem Abstand von 2-5 Nukleotiden aufzunehmen. Es besteht die Möglichkeit, mit anderen Etikettensystemen wie Megastar dye (DYXL; Dyomics, Jena, Deutschland) und Pulsar 650 (Biosearch Technologies, Inc., Novato, CA, USA), aber diese Beschriftungssysteme benötigen spektrale Informationen, um das Kanalübersprechen zu kompensieren, und sind nicht allgemein verfügbar. Die in dieser Studie entwickelte FRET-TaqMan-Sonde benötigt jedoch keine Farbkompensationssoftware und verwendet Fluorophore (FAM, Cy5.5), die weit verbreitet sind. Der vorliegende Bericht konzentriert sich auf das Design einer dreifach markierten Sonde, um Multiplexing in Instrumenten zu ermöglichen, die mit nur einer Anregungsquelle mit blauer Leuchtdiode (LED) (470 nm) und zugehörigen drei entsprechenden Detektionskanälen (z. B. 530 nm, 640 nm und 705 nm) ausgestattet sind.
Materialien und Methoden
Sondenkonstruktionsprinzip
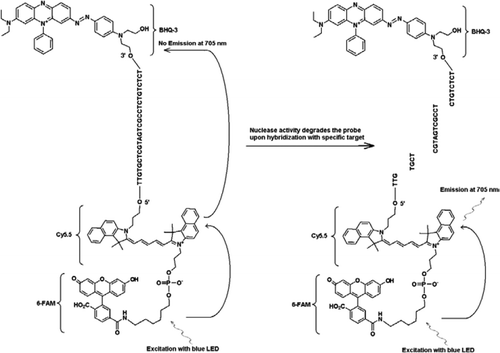
Alle Experimente wurden mit dem R.A.P.I.D.-System von Idaho Technology durchgeführt, einem tragbaren Echtzeit-PCR-Instrument mit 32 Proben, das mit einer blauen LED zur Anregung (470 nm) und den zugehörigen drei Fluoreszenzdetektionskanälen ausgestattet ist, die die Fluoreszenz bei 530, 640 und 705 nm für mono- und zweifarbige Assays messen. Die Primer JVAF und JVAR (10) und die Sonde 5′-FAM/ BHQ-1 wurden in der CDC Biotechnology Core Facility (Atlanta, GA, USA) synthetisiert, und die Sonde 5’FAM-Cy5.5/3’BHQ-3 wurde bei Idaho Technology synthetisiert. Um die Duplex-TaqMan-PCR zu ermöglichen, wurde eine Sonde mit 5′-FAM/ BHQ-1 (520 nm Fluoreszenzwellenlänge) und eine andere mit 5’FAM-Cy5.5/3’BHQ-3 (705 nm Fluoreszenzwellenlänge) markiert. In Lösung bleibt die mit 5’FAM-Cy5.5/3’BHQ-3 markierte Sonde bei Hybridisierung zu einer Sequenz gequencht, und bei nachfolgender Spaltung der Sonde durch enzymatische Primerverlängerung wird der Quencher eliminiert. Dies führt zu FRET durch die Anregung von FAM durch die Lichtquelle des R.A.P.I.D. Instrument. Die Anregungsenergie wird auf den Akzeptor-Fluorophor Cy5 übertragen.5, und die emittierte Fluoreszenz wird in einem zweiten Fluoreszenzkanal (Fluoreszenzkanal F3 für das R.A.P.I.D.-Instrument) gemessen. Die Emission von Cy5.5-Fluoreszenz ist das Ergebnis der Kombination von FRET- und TaqMan-Sondentechniken, so dass diese dreifach markierten Sonden als „FRET-TaqMan“ -Sonden bezeichnet werden können.
Primer, Sonden und Plasmid
Um die Multiplexfähigkeiten des FRET-TaqMan-Designs zu demonstrieren, verwendeten wir einen gattungsspezifischen Cryptosporidium-Echtzeit-PCR-Assay, um einen 159-bp-Teil des 18S-rRNA-Gens zu amplifizieren (10). Der Vorwärtsprimer (JVAF) war 5′-ATGACGGGTAACGGGGAAT-3′, der Rückwärtsprimer (JVAR) war 5′-CCAATTACAAAAC-CAAAAAGTCC-3′ und die TaqMan-Sonde (JVAP) war 5′-FAM-CGCGCCTGCT-GCCTTCCTTAGATG–BHQ1-3′, entsprechend den Nukleotiden 100-118, 258-236, und 184-161 der GenBank accession No. AY458612. Primer für den Aufbau des Cryptosporidium IPC enthielten den gleichen Satz von Primersequenzen, die für die Amplifikation von Cryptosporidium spp., mit Ausnahme der Sondenbindungsregion der Ziel-DNA, die durch die künstliche Sequenz 5′-TTGTGCTCGTAGTCGCCTCTCTGTCTCT-3′ (JPIPC) ersetzt wurde. Diese Sondensequenz hatte eine Glühtemperatur von 69°C und war so ausgelegt, dass sie keine Sekundärstruktur aufwies. Die theoretische Spezifität für die interne Sonde wurde durch eine BLAST (11) -Suche der Sonde und der Primersequenzen ermittelt, bei der keine genaue Übereinstimmung mit einer Cryptosporidium-Sequenz gefunden wurde. Das IPC-Fragment wurde durch Integrated DNA Technologies (Coralville, IA, USA) synthetisiert und in ein Plasmid kloniert. Eine bekannte Plasmid-DNA-Konzentration (100 Kopien/Reaktion) wurde in den Mastermix aller Reaktionen außer in der Negativkontrolle gegeben. Eine für den IPC spezifische dreifach markierte Sonde (JPIPC) wurde von Idaho Technology synthetisiert, wie in Abbildung 1 gezeigt. Die FAM/Cy5.5/BHQ3 Oligonukleotidsonde wurde beginnend am 3′-Ende mit nicht fluoreszierendem Black Hole Quencher (BHQ-3) Controlled Pore Glass (CPG) synthetisiert. Das 5′-Ende wurde dann mit einem Cy5.5-Phosporamidit konjugiert, gefolgt von einem terminalen 5′-Fluorescein (6-FAM) -Phosporamidit.
Echtzeit-PCR-Assay
Durchflusszytometrie sortierte Cryptosporidium parvum-Oozysten (200 Oozysten pro Röhrchen), die vom State Laboratory of Hygiene der University of Wisconsin (Madison, WI, USA) erhalten wurden. DNA von C. parvum-Oozysten wurden unter Verwendung eines von Hill et al. (12) und suspendiert in 80 µL Tris EDTA (TE, pH 8,0) Puffer. Pro Reaktion wurden zwei Mikroliter DNA zugegeben, was 5 Oozysten entsprach (entspricht 100 Kopien der Ziel-DNA). Das Amplifikationsreaktionsgemisch bestand aus Quantifast Probe PCR + ROX Kit Reaktionsgemisch (Kat. 204354; Qiagen) ohne ROX-Zusatz, JVAF, JVAR-Primer (je 0,4 µM), JVAP-Sonde (0,2 µM), JPIPC-Sonde (0,2 µM) und 100 Kopien des IPC. Ein Aliquot (2 µL) der extrahierten DNA-Probe wurde in die Reaktionskapillare (Roche Diagnostics, Indianapolis, IN, USA) mit 18 µL Reaktionsgemisch gegeben. Eine C. parvum-genomische DNA-Positivkontrolle (Abbildung 2, Plot 1), das Plasmid IPC (Abbildung 2, Plot 2), eine Mischung aus der C. parvum-Positivkontrolle und der IPC (Abbildung 2, Plot 3) und eine No-Template-Kontrolle (NTC) bestehend aus nukleasefreiem Wasser (Kat. Nein. AM9932; Ambion, Foster City, CA, USA) anstelle der DNA (Abbildung 2, Diagramm 4) wurden in jedes Experiment einbezogen. Die Fertigstellung des Protokolls dauerte ∼30 min mit den folgenden PCR-Bedingungen: heißstart-Denaturierungsschritt bei 95 ° C für 3 min, gefolgt von 45 Zyklen mit einer 95 ° C-Denaturierung für 5 s, 60 ° C-Annealing für 30 s mit einmaliger Fluoreszenzaufnahme (mit einer Temperaturrampenrate von 20 ° C / s). Ein positives Ergebnis wurde für FAM in Kanal F1 (530 nm) und für Cy5.5 in Kanal F3 (705 nm) aufgezeichnet. Amplifikationsreaktionen wurden in dreifacher Ausfertigung durchgeführt. Amplikons (159 bp) wurden durch Agarosegelelektrophorese und glühgrüne Nukleinsäurefärbung (eEnzyme, Gaithersburg, MD, USA) sichtbar gemacht, um die Spezifität des PCR-Produkts zu bestätigen.
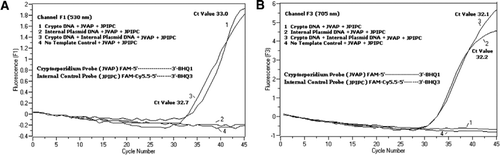
In allen PCR-Reaktionsgemischen wurde eine äquimolare Mischung aus einem FAM/BHQ1 und einem FAM/Cy5.5/BHQ3 zugegeben. (A) Kanal F1 überwacht die Zunahme der FAM-Fluoreszenz auf das Vorhandensein von C. parvum. Plot 1 (Kanal F1 sammelt hauptsächlich FAM-Signal), das nur C. parvum DNA enthielt, zeigte Cryptosporidium-spezifisches FAM-Signal in der PCR. Plot 2 enthielt nur Plasmid-DNA und kein FAM-Signal von der FAM / Cy5.5 / BHQ3-Sonde. Plot 3 enthielt beide C. parvum- und Plasmid-DNA produzierten aber nur C. parvum-spezifisches FAM-Signal. Plot 4 war eine negative Probe, die nur nukleasefreies Wasser anstelle von Template-DNA enthielt; Von den Sonden wurde keine Fluoreszenz beobachtet. (B) Kanal F3 überwacht den Anstieg von Cy5.5 auf das Vorhandensein von interner Kontrollplasmid-DNA. Plot 1 enthielt nur C. parvum DNA und kein Fluoreszenzsignal von FAM/BHQ1 Sonde. Plot 2 (Kanal F3 sammelt hauptsächlich Cy5.5–Signal), das nur Plasmid-DNA enthält, ergab ein internes Plasmid-Kontroll (IPC) -spezifisches Cy5.5-Signal in der PCR. Plot 3 enthielt beide C. parvum- und Plasmid-DNA produzierten aber nur IPC-spezifisches Cy5.5-Signal. Plot 4 war eine negative Probe, die nur nukleasefreies Wasser anstelle von Template-DNA enthielt; Von den Sonden wurde keine Fluoreszenz beobachtet.
Ergebnisse und Diskussion
Echtzeit-PCR-Fluoreszenzsignale, die die Amplifikation der genomischen C. parvum-DNA reflektieren, wurden im F1-Kanal (Abbildung 2A) aufgrund der FAM-Markierung der TaqMan-Sonde nachgewiesen. Fluoreszenzsignal, das die Verstärkung des IPC reflektiert, wurde im F3-Kanal (Abbildung 2B) aufgrund von FAM / Cy5 nachgewiesen.5 beschriftung der IPC TaqMan Sonde. Diese Sonde erzeugte einen Anstieg von Cy5.5 im F3-Kanal (Cy5.5), aber keinen Anstieg von FAM im F1-Kanal (FAM), wenn BHQ3 aufgrund der Nukleaseaktivität vom FAM / Cy5.5 getrennt wird, da Energie von FAM auf Cy5.5 übertragen wurde. In ähnlicher Weise wurde bei der Duplexreaktion die genomische DNA-Amplifikation von C. parvum im F1-Kanal nachgewiesen, während die IPC-Amplifikation gleichzeitig im F3-Kanal nachgewiesen wurde. FAM hat eine Anregung bei 495 nm und eine Emission bei 520 nm. Cy5.5 hat eine Anregung von 690 nm und eine Emission von 705 nm. Bei einer solchen Nähe zwischen Cy5.5 und FAM auf der dreifach markierten IPC-Sonde emittiert der Cy5.5-Fluorophor Fluoreszenz aufgrund der vollständigen Energieübertragung vom FAM-Fluorophor, was zu keiner Fluoreszenzemission von FAM im F1-Kanal führt (Abbildung 2A). Negative Kontrollreaktionen zeigten keinen Anstieg der Fluoreszenz aufgrund von FAM oder Cy5.5. Diese Ergebnisse zeigen auch, dass eine einzelne blaue (470 nm) LED-Quelle ausreicht, um doppelte Fluoreszenzemissionen durch F1- (530 nm) und F3- (705 nm) Kanäle ohne spektrale Überlappung in den jeweiligen Überwachungskanälen für FAM und Cy5.5 zu überwachen. Die FRET-TaqMan-Technik wurde auch unter Verwendung eines Panels von 12 DNA-Extrakten aus Stuhlproben getestet (8 aus Proben, die zuvor als positiv für Cryptosporidium spp. und vier Proben, die negativ auf Cryptosporidium spp.). Der 18S-rRNA-Assay konnte alle acht Cryptosporidium spp. Es wurde festgestellt, dass der IPC-TaqMan-Assay nur die IPC-Plasmid-DNA amplifiziert. Der IPC-Assay ergab Ct-Werte von 32-37, was auf bis zu 5 Ct-Werte der PCR-Hemmung für einige Stuhlproben hindeutete (32 war der erwartete Ct-Wert für No-Inhibitionsreaktionen).
Die vorliegende Technik unterscheidet sich von traditionell verwendetem FRET dadurch, dass anstelle von zwei Sonden (FRET-Sonden) eine fluoreszenzmarkierte Oligonukleotidsonde (FRET-TaqMan-Sonde) verwendet wird, bei der eine Sonde den Donorfluorophor und die andere Sonde den Akzeptorfluorophor enthält (13,14). Die FRET-TaqMan-Sonde ist am 5′-Ende mit zwei Fluorophoren von Donor- (FAM) und Akzeptorfluorophoren (Cy5.5) und am 3′-Ende mit einem BHQ-3-Quencher wie einer TaqMan-Sonde markiert. Die einzigartige Sondenbindungsregion wird ausgewählt, um sich von der Ziel-DNA zu unterscheiden, um zwei verschiedene Fluoreszenzmessungen in zwei verschiedenen Detektionskanälen (530 nm in F1 und 705 nm in F3) aus derselben Amplifikationsreaktion zu überwachen, ohne dass eine Farbkompensationssoftware enthalten sein muss. Die Ziel-DNA und IPC hatten die gleichen Primer-Bindungsstellen, aber eine andere Sondensequenz. Dies stellt sicher, dass die Verwendung der gleichen Primer für beide DNA-Targets Primer-Dimer-Bildung oder Interaktion zwischen Primer-Sets zu minimieren. Wenn FAM / Cy5.5-Fluorophore nahe am 5′-Ende platziert werden, findet ein Resonanzenergietransfer mit einer Zunahme der Fluoreszenzemission des Cy5.5-Fluorophors statt. Die Verwendung von BHQ3, einem Nicht-Fluoreszenzakzeptor für Cy5.5, verringert jedoch die potenzielle Hintergrundfluoreszenz, was zu einem höheren Signal-Rausch-Verhältnis führt. Es wurde gefunden, dass Zugabe von 100 Kopien des IPC zu einer Reaktion enthaltend ∼100 Kopien von genomischem C. parvum DNA ergab ähnliche Ct-Werte (Ct = 32), wenn der Cryptosporidium TaqMan Assay als Singleplex Assay und als Duplex Assay mit dem IPC TaqMan Assay durchgeführt wurde. Bei Anwendung auf eine Gruppe von Cryptosporidium-positiven DNA-Proben zeigte der IPC, dass einige Proben keine PCR-Inhibitoren enthielten (basierend auf erwarteten Ct-Werten von 32), andere Proben jedoch mit einer Hemmung von bis zu 5 Ct-Werten assoziiert waren.
Die neu entwickelte interne PCR-Positivkontrolle erwies sich aufgrund der Markierung mit dem FAM/Cy5.5-BHQ3 und FAM/BHQ1 für das jeweilige IPC- und 18S-rRNA-Zielgen in einer Dual-Color-Multiplex-PCR zur Überwachung in F1- und F3-Fluoreszenzkanälen des R.A.P.I.D-Instruments als sensitiv und spezifisch für den gleichzeitigen Nachweis von zwei Targets in derselben Reaktion. Die Verwendung der 5’FAM-Cy5.5/3’BHQ-3–markierten Sonde als zweiter Reporter ist vorteilhaft, da die Emissionsmaxima der beiden Sonden weit voneinander beabstandet waren. Das FRET-TaqMan Triple-markierte Sondendesign könnte Anwendungen bei der Entwicklung eines kostengünstigen Einweg- „Lab-on-a-Chip“ -Geräts mit einer Optik haben, die aus einer blauen LED-Anregung und zwei Emissionsquellen von 530 nm und 705 nm besteht, mit geeigneten optischen Filtern zum Nachweis von FAM in einem Kanal und Cy5.5 im anderen. Diese Kombination würde keine zwei Hochleistungs-Fluoreszenzquellen- und -detektionssysteme erfordern.
Danksagung
Die Autoren danken Mike Powers von der DNA Sequencing and Genomic Core Facility (University of Utah, Salt Lake City, UT, USA) und Rachelle Muller von Idaho Technology, Inc. zur Diskussion über die Entwicklung der internen Positivkontrolle für den R.A.P.I.D Cycler. Diese Veröffentlichung wurde teilweise durch Mittel unterstützt, die von den Zentren für die Kontrolle und Prävention von Krankheiten, dem Koordinierungsbüro für Terrorismusvorsorge und Notfallmaßnahmen, zur Verfügung gestellt wurden. Die Verwendung von Handelsnamen und kommerziellen Quellen dient nur zur Identifizierung und impliziert keine Billigung durch die Zentren für die Kontrolle und Prävention von Krankheiten oder das US-Gesundheitsministerium.
- 1. Hartman, L.J., S.R. Coyne und D.A. Norwood. 2005. Entwicklung einer neuartigen internen Positivkontrolle für Taqman-basierte Assays. Mol. Zelle. Matthäus 19:51-59.Querverweis, Medline, CAS, Google Scholar
- 2. Hoorfar, J., B. Malorny, A. Abdulmawjood, N. Cook, M. Wagner, und P. Fach. 2004. Praktische Überlegungen zum Entwurf interner Amplifikationskontrollen für diagnostische PCR-Assays. J. Clin. Microbiol. 42:1863–1868.Querverweis, Medline, CAS, Google Scholar
- 3. Betsou, F., K. Beaumont, J.M. Sueur, und J. Orfila. 2003. Konstruktion und Auswertung von interner Kontroll-DNA zur PCR-Amplifikation von Chlamydia trachomatis DNA aus Urinproben. J. Clin. Microbiol. 41:1274–1276.Querverweis, Medline, CAS, Google Scholar
- 4. Burggraf, S. und B. Olgemoller. 2004. Einfache Technik zur internen Kontrolle von Echtzeit-Amplifikationstests. Clin. Chem. 50:819–825.Querverweis, Medline, CAS, Google Scholar
- 5. Drosten, C., M. Weber, E. Seifried, und W.K. Roth. 2000. Bewertung eines neuen PCR-Assays mit kompetitiver interner Kontrollsequenz für das Blutspenderscreening. Transfusion 40:718-724.Querverweis, Medline, CAS, Google Scholar
- 6. Gruber, F., F.G. Falkner, F. Dorner, und T. Hammerle. 2001. Quantifizierung von viraler DNA durch Echtzeit-PCR unter Anwendung von Duplexamplifikation, interner Standardisierung und zweifarbigem Fluoreszenznachweis. Appl. ENVIRON. Microbiol. 67:2837–2839.Crossref, Medline, CAS, Google Scholar
- 7. Rosenstrauß, M., Z. Wang, S.Y. Chang, D. DeBonville und J.P. Spadoro. 1998. Eine interne Kontrolle für routinediagnostische PCR: Design, Eigenschaften und Auswirkungen auf die klinische Leistung. J. Clin. Microbiol. 36:191–197.Medline, CAS, Google Scholar
- 8. Stocher, M., V. Leb, und J. Berg. 2003. Ein praktischer Ansatz zur Generierung mehrerer interner Kontroll-DNA für ein Panel von Echtzeit-PCR-Assays. In: J. Virol. Methoden 108:1-8.Querverweis, Medline, CAS, Google Scholar
- 9. Zimmermann, K. und J.W. Mannhalter. 1996. Technische Aspekte der quantitativen kompetitiven PCR. Biotechniken 21:268-279.Link, CAS, Google Scholar
- 10. Jothikumar, N., A.J. da Silva, I. Moura, Y. Qvarnstrom und V.R. Hill. 2008. Nachweis und Differenzierung von Cryptosporidium hominis und Cryptosporidium parvum durch Dual-TaqMan-Assays. Dr. Med. Microbiol. 57:1099–1105.Crossref, Medline, CAS, Google Scholar
- 11. Altschul, S.F., W. Gish, W. Miller, E.W. Myers und D.J. Lipman. 1990. Grundlegende lokale Ausrichtung Suchwerkzeug. In: J. Mol. Biol. 215:403–410.Crossref, Medline, CAS, Google Scholar
- 12. Hill, V.R., A.M. Kahler, N. Jothikumar, T.B. Johnson, D. Hahn und T.L. Cromeans. 2007. Multistate Evaluation eines ultrafiltrationsbasierten Verfahrens zur gleichzeitigen Rückgewinnung von magensaftresistenten Mikroben in 100-Liter-Leitungswasserproben. Appl. ENVIRON. Microbiol. 73:4218–4225.Crossref, Medline, CAS, Google Scholar
- 13. Bernard, P.S., M.J. Lay und C.T. Wittwer. 1998. Integrierte Amplifikation und Detektion der C677T-Punktmutation im Methylentetrahydrofolatreduktase-Gen durch Fluoreszenzresonanzenergietransfer und Sondenschmelzkurven. Anal. Biochem. 255:101–107.Crossref, Medline, CAS, Google Scholar
- 14. Wittwer, C.T., M.G. Herrmann, A.A. Moss und R.P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130–138.Link, CAS, Google Scholar