Słaby w kolanach: zespół Millera Fishera
zespół Millera Fishera (MFS), rzadka, samoograniczająca się odmiana zespołu Guillaina-Barrégo (GBS), jest zespołem przeciwciał anty-GQ1bIgG, który wpływa na obwodowy i ośrodkowy układ nerwowy.1 ostra polineuropatia nerwowo-mięśniowa, MFS powoduje paraliż wstępujący z klasyczną triadą prezentacji oftalmoplegia, ataksja i arefleksja. Naukowcy po raz pierwszy opisali triadę kliniczną w 1932 roku.1 dwie dekady później kanadyjski Specjalista ds. udaru mózgu Charles Miller Fisher jako pierwszy opublikował w 1956 r. raport, w którym opisał wyniki kliniczne i zdefiniował stan jako ograniczoną formę GBS.2
objawy występują w ciągu kilku dni, często zimą i wiosną, zazwyczaj po infekcji wirusowej, a pacjenci często doświadczają podwójnego widzenia jako jednego z pierwszych objawów. Kiedy pacjent ma objawy takie jak przerywana ezotropia, zaburzenia czynności poziomych i stałe źrenice źrenic, a badanie neurologiczne jest nieprawidłowe, czas pomyśleć o GBS, a konkretnie MFS. Poniższy przypadek ilustruje istotne objawy przedmiotowe i podmiotowe oraz odzwierciedla typowy wynik.
Historia
27-letni mężczyzna rasy kaukaskiej przedstawiony klinice okulistycznej VA w miesiącach wiosennych zgłaszający ostre podwójne widzenie obuoczne, które rozpoczęło się dzień wcześniej. Pacjent stwierdził, że był stały, ale występował tylko przy oglądaniu na odległość z obiektami przesuniętymi poziomo. Nie stwierdzono bólu oka ani niewyraźnego widzenia związanego z podwójnym widzeniem. Skarżył się również na niedawne zawroty głowy i problemy z równowagą. Zgłosił infekcję zatok ze znacznym przekrwieniem przynosowym, które zaczęło się tydzień przed podwójnym widzeniem. Wcześniejsze historie okulistyczne i medyczne były nijakie; pacjent nie przyjmował obecnie żadnych leków i nie miał alergii na leki.
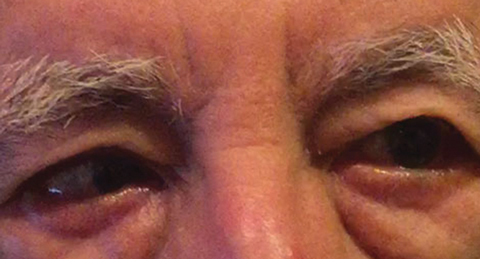
rys. 1. Diagnostyka różnicowa po badaniu wyjściowym obejmowała częściowe porażenie CN VI prawej strony, które może wskazywać na stan naczyniowy, jak u innego pacjenta. Kliknij obraz, aby powiększyć. Zdjęcie: Michael DelGiodice, OD
dane diagnostyczne
podczas badania podstawowego, nieskorygowane ostrości wizualne wynosiły 20/20-2 OD i 20/25-2 OS. Źrenice były 5 mm jednakowo okrągłe i reagowały na światło bez defektu źrenic aferentnych. Ruchy pozajelitowe były pełne, gładkie i dokładne w obu oczach, bez żadnych ograniczeń. Test pokrycia na odległość ujawnił przerywaną od czterech do sześciu pryzmatycznych dioptrii prawej ezotropii w spojrzeniu pierwotnym występującą w około 50% czasu. Przeprowadzając test okrywowy w near ujawniono cztery pryzmatyczne ezoforie dioptrii w spojrzeniu pierwotnym bez tropii.
chociaż w tym przypadku nie jest wykonywana, klinicyści powinni wykonać test w pierwszym, prawym I lewym spojrzeniu. U pacjentów z podwójnym wzrokiem porażenie nerwu czaszkowego szóstego (CN VI) objawia się jako odchylenie niekomitacyjne, gorsze w spojrzeniu prawym lub lewym, w zależności od tego, po której stronie znajduje się porażenie. Z drugiej strony dekompensująca się Foria wykaże odchyłkę komitywną.
błąd refrakcji wynosił -0,50 d sfery OD I -0,75 D sfery OS i widzenie było korygowane do 20/20 w każdym oku. Ciśnienie wewnątrzgałkowe w obu oczach mieściło się w granicach normy. Wyniki przedniego i tylnego odcinka nie były widoczne w prawym I lewym oku. Nerwy wzrokowe wyglądały na zdrowe z proporcją filiżanki do tarczy 0,15 W obu oczach.
diagnostyka różnicowa obejmowała częściowe porażenie CN VI prawej strony (ryc. 1), miastenię, proces ściskania i zdekompensowaną ezoforię odległości powodującą przerywaną ezotropię prawej strony. Pacjent zaprzeczył istnieniu dwuocznego widzenia w wywiadzie.
obrazowanie mózgu i Orbit zostało zlecone przez klinikę okulistyczną w celu wykluczenia zaangażowania mózgu, w tym zmiany uciskowej. Tomografia komputerowa (CT) skany mózgu i Orbit zostały wykonane i zinterpretowane przez neuroradiologa personelu. CT wybrano zamiast rezonansu magnetycznego (MRI) ze względu na czas i dostępność; badania i wyniki można było uzyskać tego samego dnia z CT w klinice po godzinach. Wyniki były nijakie, bez oznak krwotoku wewnątrzczaszkowego,zmiany uciskowej lub ostrego zawału.
tomografia komputerowa zatok przynosowych wykazała umiarkowaną do ciężkiej przewlekłą chorobę zatok przynosowych i całkowite zmętnienie prawej zatoki szczękowej. Nie było przedłużenia orbity. Lekarz rodzinny pacjenta został poinformowany o objawach pacjenta, wynikach badania oczu i wynikach obrazowania tomografii komputerowej i rozpoczął leczenie zapalenia zatok szczękowych doustnym pięciodniowym zestawem dawek azytromycyny i zalecił pacjentowi kontynuację leczenia w klinice okulistycznej.
pacjent wrócił pięć dni później po zakończeniu doustnego kursu azytromycyny i stwierdził, że nie odczuwa poprawy w diplopii na odległość; ponadto doświadczył wystąpienia istotnej światłowstręt i bólu okołookularowego wokół obu oczu. Pacjent stwierdził, że pogarsza się również jego nierównowaga chodu i trudności z chodzeniem. Podczas badania, najlepiej skorygowane ostrości wzroku pozostały 20/20 w prawym I lewym oku. Źrenice rozszerzono na 8,5 mm bez reakcji na światło w każdym oku (ryc. 2). Ruchy pozajelitowe ujawniły boczne ograniczenie z bólem podczas ruchu w prawym oku i przyśrodkowe ograniczenie z bólem podczas ruchu w lewym oku zgodne z porażeniem prawego spojrzenia (ryc. 3). Zajęcia poziome i pionowe były saccadic z niedokładnymi i nierównymi ruchami. Wyniki segmentu przedniego i tylnego pozostały nijakie.
diagnostyka różnicowa po badaniu okulistycznym obejmowała lewostronną oftalmoplegię śródjądrową, porażenie prawojądrowe i porażenie prawojądrowe z lewym oftalmoplegią śródjądrową, co może odpowiadać za zmiany ruchliwości pozajądrowej. Inne różnice obejmują uszkodzenie śródmózgowia lub miastenię. Diagnostyka różnicowa rozszerzania źrenic obejmuje porażenie nerwu trzeciego (CN III), uraz i rozszerzenie farmakologiczne. U pacjenta nie stwierdzono innych objawów wskazujących na porażenie nerwu trzeciego i nie stwierdzono u niego urazu ani narażenia na działanie środków farmakologicznych.
pacjent został skierowany do neurologii w celu dalszej oceny. Badanie neurologiczne wykazało, że pacjent był w pełni czujny i zorientowany bez utraty czucia twarzy. Pacjent wykazywał łagodną ataksję z trudnością chodzenia w tandemie i stania z pozycji kucającej. Występowały bliższe osłabienie kończyn górnych i dolnych oraz nieprawidłowe odruchy. Wszystkie zmysły były nienaruszone. MRI i angiografia rezonansu magnetycznego (MRA) mózgu i Orbit były nijakie. Białko płynu mózgowo-rdzeniowego było podwyższone o 75 mg/dL (zakres odniesienia: 15 mg/dL do 45 mg / dL) bez innych nieprawidłowości. Badanie elektromiogramem było mało znaczące i nie wykazało żadnych nieprawidłowości w przewodnictwie nerwowym od kręgosłupa do stóp i dłoni.
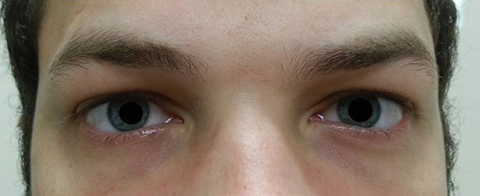
rys. 2. Źrenice pacjenta były rozszerzone o 8,5 mm bez reaktywności w prawym lub lewym oku. Kliknij obraz, aby powiększyć.
rozpoznanie
u pacjenta wystąpiły ostre objawy podwójne widzenie, ataksja i arefleksja. Biorąc pod uwagę przerywaną ezotropię prawa, upośledzone zajęcia poziome, naprawioną źrenicę mydriatyczną, łagodną ataksję i nieprawidłowe odruchy na egzaminie neurologicznym i podwyższone białko płynu mózgowo-rdzeniowego, postawiono diagnozę MFS. Pacjent został przyjęty do szpitala w celu monitorowania dalszych powikłań. Pacjentowi dostarczono opaskę na oko w celu zakrycia prawego oka w razie potrzeby podwójnego widzenia.
obecnie nie ma opublikowanych kryteriów diagnostycznych dla zespołu Millera Fishera. Diagnoza jest zwykle dokonywana poprzez prezentację triady klinicznej wraz z obrazowaniem i badaniami płynu mózgowo-rdzeniowego. MRI jest zwykle nijakie w tym stanie, chociaż niektóre badania wykazały Centralne zmiany w śródmózgowiu, pons i dolnym rdzeniu. Badania elektrofizjologiczne wykazują zmniejszone lub nieprawidłowe przewodnictwo czuciowe obwodowe.
białko płynu mózgowo-rdzeniowego jest często podwyższone bez innych nieprawidłowych wyników. W przełomowym badaniu u 64,4% pacjentów z MFS stwierdzono podwyższone stężenie białka płynu mózgowo-rdzeniowego.Pozytywny wynik testu przeciwciała anty-Gq1b IgG pozwala na ostateczne rozpoznanie. W jednym badaniu przeciwciała anty-Gq1b IgG występowały u maksymalnie 95% pacjentów z tą chorobą i nie występowały w grupie kontrolnej.3
neurolog zdiagnozował u pacjenta MFS na podstawie klinicznej triady oftalmoplegii, ataksji i arefleksji, nijakiego MRI i MRA mózgu oraz podwyższonego białka płynu mózgowo-rdzeniowego. W związku z tym w tym przypadku nie przeprowadzono badań na obecność przeciwciał anty-Gq1b IgG.
kilka cech klinicznych MFS jest również widoczne w zespole Guillain-Barré i zapaleniu mózgu pnia mózgu Bickerstaffa, co sprawia, że diagnoza jest trudna. U pacjentów z tylko Guillain-Barré występują osłabienie kończyn, utrata czucia, neuropatia czaszkowa i arefleksja bez objawów oftalmologicznych. Pacjenci z zapaleniem mózgu pnia mózgu Bickerstaffa zazwyczaj występują z oftalmoplegią, ataksją, hiperrefleksją i zaburzoną świadomością. Ponieważ kilka objawy i oznaki są obecne we wszystkich trzech warunkach, anty-Gq1b IgG miana przeciwciał testowanie jest pomocne dla bardziej ostatecznej diagnozy. Przeciwciała anty-Gq1b IgG są dodatnie u znacznej większości pacjentów z zespołem Millera Fishera w porównaniu z 66% pacjentów z zapaleniem mózgu pnia mózgu Bickerstaffa i 26% pacjentów z zespołem Guillaina-Barrégo.16
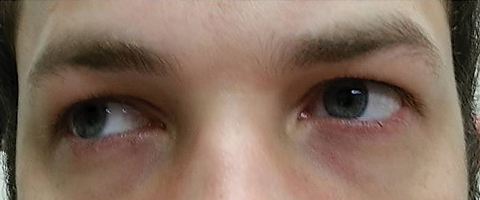
rys. 3. Ruchy pozajelitowe ujawniły ograniczenie dekstrowersji. Kliknij obraz, aby powiększyć.
leczenie i obserwacja
pacjent został oceniony dzień po przyjęciu, kiedy stwierdził, że nie doświadczył zmian wzrokowych ani poprawy równowagi. Podwójne widzenie obuoczne było nadal stałe, dlatego prawe oko pozostało połatane. Ból oczu z dalekimi spojrzeniami i szybkimi ruchami oczu były nadal obecne. Przy ograniczonym badaniu przy łóżku najlepiej skorygowane ostrości wzroku pozostały 20/20 w prawym I lewym oku. Źrenice zostały rozszerzone o 8.0mm w obu oczach przy minimalnej reaktywności, która była nieco lepsza od poprzedniego dnia. Widzenie kolorów było normalne w prawym I lewym oku. Ruchy pozajelitowe były pełne we wszystkich ćwiartkach z bólem wciąż obecnym na dalekich horyzontalnych spojrzeniach. Zajęcia poziome pozostały sakkadyczne z ruchami szarpanymi i oczopląsem końcowym. Nerwy wzrokowe wyglądały na zdrowe z wyraźnymi marginesami i stosunkiem filiżanki do krążka wynoszącym 0,15 W obu oczach.
dwa dni później pacjent stwierdził poprawę wszystkich objawów i że jego oczy „czują się dziś lepiej niż od jakiegoś czasu.”Ból oka i podwójne widzenie obuoczne ustąpiły. Ostrość widzenia pozostała 20/20 w prawym I lewym oku. Rozmiary źrenic wynosiły 7,5 mm w kolorze ciemnym i 7,0 mm w świetle, z wyraźną i wyraźną odpowiedzią w obu oczach. Ruchy pozajelitowe były pełne we wszystkich ćwiartkach bez bólu oka. Zajęcia poziome pozostały sakkadyczne z ruchami szarpanymi i oczopląsem końcowym. Badanie neurologiczne wykazało nieprawidłowe odruchy, które pozostały stabilne. Opcje leczenia MFS obejmują plazmaferezę i dożylną (IV) immunoglobulinę IgG, ale te zostały wstrzymane ze względu na poprawę w oftalmoplegii i ataksji.
cztery dni później pacjent zgłosił, że wszystkie objawy nadal się poprawiają, z pewnymi trudnościami z ogniskowaniem i śledzeniem podczas korzystania z komputera. Równowaga była w 80% rozwiązana i niewiele było przypadków diplopii na odległość. Acuities pozostał 20/20 OU. Źrenice miały 5,5 mm W ciemności i 4,0 mm w świetle z 3 + bezpośrednimi i zgodnymi odpowiedziami. Ruchy pozajelitowe były pełne we wszystkich ćwiartkach bez bólu oka. Ulepszono prowadzenie poziome, bez ruchów sakkadowych. Zakwaterowanie monokularowe było w normie, mierzono odpowiednio 8,5 i 9 dioptrii pryzmatycznych OD I OS.
tydzień później pacjent zgłosił, że jego widzenie wróciło do normy bez podwójnego widzenia i łagodnych trudności z ogniskowaniem i śledzeniem. Równowaga była w 100% rozwiązana. Ostrość widzenia pozostała 20/20 w prawym I lewym oku. Rozmiary źrenic wynosiły 5,5 mm W ciemności, 4,0 mm w świetle z 4 + bezpośrednimi i zgodnymi odpowiedziami. Ruchy pozajelitowe były pełne we wszystkich ćwiartkach bez bólu oka. Ćwiczenia poziome poprawiły się bez zaobserwowanych ruchów sakkadowych. Oczne zdrowie było nijakie OU.
leczenie wstrzymano ze względu na ustąpienie oftalmoplegii i ataksji ze stabilną arefleksją. Całkowite wyleczenie okulistyki i ataksji zajęło dwa tygodnie. Leczenie obejmowało ścisłe monitorowanie i obserwację w ciągu czterech tygodni; pacjentowi zalecono wcześniejszy powrót, jeśli którykolwiek z objawów powróci.
cechy kliniczne
w dobrze opisanych badaniach MFS u wszystkich pacjentów stwierdzono triadę kliniczną oftalmoplegii, ataksji i arefleksji.4, 5 początek objawów zwykle występuje w ciągu kilku dni, a pacjenci cierpią na infekcję wirusową na jeden do czterech tygodni przed wystąpieniem objawów klinicznych.4,5 według jednego badania z udziałem 50 pacjentów z tą chorobą średni odstęp między początkiem zakażenia a rozwojem objawów neurologicznych wynosi osiem dni.Inne objawy MFS obejmują niewyraźną mowę, trudności w połykaniu i nieprawidłowy wyraz twarzy z niezdolnością do uśmiechu lub gwizdania.
cechy okulistyczne. Lekarze powinni zwracać uwagę na rozszerzenie źrenicy, cofnięcie powiek, ostre zamknięcie kąta i podwójne widzenie wtórne do porażeń nerwowych dotykających CN III, IV i VI. 3,8,10,22,24 podwójne widzenie jest najczęstszym objawem okulistycznym zgłaszanym w wielu badaniach MFS.8,16,25,26,27 podwójne widzenie jest również początkowym objawem zgłaszanym u 38,6% z 223 pacjentów w jednym przełomowym badaniu i u 65% z 466 pacjentów w drugim badaniu.8,27 w drugim badaniu u 100% pacjentów wystąpiła oftalmoplegia zewnętrzna, natomiast oftalmoplegia wewnętrzna u 35% pacjentów.27 oftalmoplegia zewnętrzna odnosi się do upośledzenia mięśni zewnętrznych zewnątrzgałkowych. Wewnętrzna oftalmoplegia odnosi się do upośledzenia zwieracza źrenic i mięśni rzęskowych. Całkowita ophthalmoplegia wpływa zarówno na mięśnie zewnętrzne, jak i wewnętrzne.
w retrospektywnym badaniu z udziałem 19 pacjentów z MFS, u wszystkich stwierdzono jedno lub więcej porażeń nerwowych wpływających na mięśnie zewnątrzgałkowe.Inne badania wykazały, że u pacjentów z MFS może wystąpić wiele porażeń nerwów czaszkowych, które mogą objawiać się obustronnie.22,24,25 jednym z częstych stwierdzeń występujących wśród dodatnich zaburzeń IgG anty-Gq1b jest deficyt uprowadzenia zgodny z jednostronnym lub obustronnym porażeniem CN VI.26,28 oftalmoplegia wewnętrzna powoduje rozszerzenie źrenicy, gdzie zwężenie źrenicy do światła i / lub bliskiej stymulacji może wynosić od minimalnego do nieobecnego.8,26 w jednym raporcie, rozszerzenie źrenicy występowało u 42% pacjentów. Udział CN VII wystąpił u około 45,7% pacjentów z MFS, powodując osłabienie twarzy, niezdolność do uśmiechu lub gwizdania.8
ataksja. Przyczyna tej cechy klinicznej w MFS nie jest w pełni poznana. Istnieje debata na temat tego, czy ataksja jest spowodowana dysfunkcją centralnie w móżdżku, czy obwodowo. Pierwsza hipoteza, zaproponowana przez dr Fishera, postulowała udział neuronów aferujących Ia.2 zostało to poparte drugim badaniem dekady później, które wykazało nieprawidłowości włókien aferentnych Ia w MFS.Zaproponowano również nieprawidłowe zajęcie nerwów obwodowych z niedopasowaniem między proprioceptywnym wkładem z wrzecion mięśniowych a informacjami kinestetycznymi z receptorów stawowych.12
istnieją również dowody potwierdzające, że ataksja pochodzi z móżdżku. Dominację przeciwciał anty-Gq1b IgG w móżdżku stwierdzono stosując immunocytochemiczne zabarwienie ludzkiego móżdżku i dodatkowo potwierdzono badaniami western blot, które wykazały zwiększenie przeciwciał anty-Gq1b IgG u pacjentów z MFS w porównaniu z grupą kontrolną, chociaż nie badano mechanizmu zaangażowania móżdżku.13,14 w 2015 r. naukowcy opublikowali raport przypadku pacjenta z MFS, który przeszedł rezonans magnetyczny. Stwierdzono zmniejszony stosunek N-acetyloasparaginian / kreatyna, co sugeruje dysfunkcję móżdżku, która powróciła do normy 2,5 miesiąca po wyzdrowieniu.23
arefleksja. Ta cecha kliniczna, wraz z depresją głębokich odruchów ścięgnistych, jest oznaką zaangażowania obwodowego układu nerwowego w MFS. Badania elektrofizjologiczne potwierdziły nieprawidłowe przewodnictwo nerwów obwodowych w GBS i MFS.3 w jednym badaniu, 81,6% przypadków MFS z arefeksją.W innym przełomowym badaniu wykazano, że wszyscy pacjenci doświadczali arefleksji, która nadal występowała sześć miesięcy po jej wystąpieniu.4
Jak często jest MFS?
dane epidemiologiczne dotyczące zespołu Millera Fishera są ograniczone. Częstość występowania MFS jest dość rzadka, występuje u 0,09 na 100 000 osób rocznie.3 MFS występuje częściej u pacjentów z zespołem Guillain-Barré, od 3% do 25%.4,6,7 związek ten występuje częściej u pacjentów pochodzenia Dalekowschodniego, co sugeruje możliwy składnik genetyczny do MFS. Średni wiek zachorowania wynosi 43 lata, przy przedziale od 13 do 78,5 MFS dotyka mężczyzn dwa razy częściej niż kobiety, przy stosunku od 2 do 1,03,5 MFS występuje częściej w zimie i wiosną.5,6,8 dokładna przyczyna predylekcji sezonowej nie została określona, ale jest prawdopodobnie związana z pozakaźnym charakterem choroby, ponieważ stwierdzono, że infekcje bakteryjne i wirusowe wywołują odpowiedź autoimmunologiczną powodującą wytwarzanie przeciwciał anty-Gq1b IgG w MFS.
Patofizjologia
patofizjologia zespołu Millera Fishera nie jest dobrze poznana; istnieje jednak kilka hipotez z badań immunologicznych i histologicznych. Powszechnie wiadomo, że stan ten mieści się w spektrum zespołów przeciwciał anty-Gq1b IgG wraz z zespołem Guillain-Barré i zapaleniem mózgu pnia mózgu Bickerstaffa.
gangliozyd GQ1b jest grupą złożonych lipidów zaangażowanych w centralny i obwodowy układ nerwowy. GQ1b jest składnikiem struktury błony plazmatycznej w nerwach czaszkowych, które zaopatrują mięśnie zewnątrzgałkowe i bierze udział w funkcjonowaniu komórek w presynaptycznym złączu nerwowo-mięśniowym.
mechanizm autoimmunologiczny z poprzedzającej infekcji wyzwalającej wytworzy przeciwciało anty-Gq1b IgG, które uszkadza funkcję gangliozydu GQ1b, powodując demielinizację. Badania histologiczne wykazały demielinizację i obrzęk aksonalny w nerwach obwodowych i okulomotorycznych czaszki.Przeciwciało anty-GQ1bIgG jest nieobecne u zdrowych pacjentów, ale dodatnie u ponad 90% pacjentów z MFS.16,25
zakażenia bakteryjne i wirusowe wywołują odpowiedź autoimmunologiczną, powodując wytwarzanie przeciwciał anty-gq 1B IgG. Następujące czynniki zakaźne, które były związane z MFS to mycoplasma pneumonia, HIV, Campylobacter jejuni, hemophilus influenza, Helicobacter pylori i wirus Epsteina-Barr.4,16 w jednym raporcie, który dotyczył 466 pacjentów z MFS, 90% miało wcześniejszą chorobę, w tym infekcję górnych dróg oddechowych, biegunkę lub obie te choroby.
rokowanie i leczenie
zespół Millera Fishera jest chorobą samoograniczającą się i ma ogólnie pozytywne rokowanie. Objawy zwykle ustępują po kilku tygodniach, a pełne wyleczenie zwykle następuje w ciągu dwóch do trzech miesięcy. W jednym z badań poprawa zaczęła się średnio po 13 dniach od wystąpienia objawów, a całkowite ustąpienie oftalmoplegii i ataksji nastąpiło w ciągu sześciu miesięcy.4,5 nawrotów stwierdzono w mniej niż 3% przypadków.4,5
chociaż MFS jest zazwyczaj samoograniczający się, powikłania ogólnoustrojowe były związane z chorobą. Zakażenia górnych dróg oddechowych stwierdzono u 56% do 76% pacjentów z MFS, co może prowadzić do niewydolności oddechowej wymagającej wentylacji mechanicznej.Inne rzadkie poważne powikłania to kardiomiopatia, kwasica mleczanowa i śpiączka.3,20
możliwości leczenia MFS obejmują plazmaferezę, dożylną immunoglobulinę IgG i monitorowanie bez leczenia. Naukowcy postulują, że plazmafereza i IV IgG może być skuteczne do przyspieszenia czasu rozdzielczości od przeciwciała Gq1b jest IgG w klasie i okres półtrwania IgG wynosi około 21 dni, który jest dłuższy niż pięć do sześciu dni półtrwania IgM i IgA.Nie przeprowadzono jednak randomizowanych, podwójnie zaślepionych, kontrolowanych placebo badań dotyczących tych metod leczenia.
jedno retrospektywne badanie nie wykazało, że plazmafereza zmieniła szansę pełnego wyzdrowienia i czas do ustąpienia ataksji i oftalmoplegii; co więcej, odstęp od początku do rozpoczęcia leczenia plazmaferezą nie wpływał na czas do ustąpienia lub nasilenie objawów w momencie ich wystąpienia. Jednak jeden opis przypadku wskazuje, że plazmafereza jest wskazana, gdy występuje głęboka ataksja, zaburzenia czynności motorycznych i oddechowych.
pacjentów należy ściśle monitorować pod kątem ryzyka wystąpienia ciężkich powikłań układowych, takich jak niewydolność górnych dróg oddechowych. Badania nie wykazują znaczącej poprawy w czasie odzyskiwania z IV IgG lub plazmaferezy leczenia w MFS, więc monitorowanie może być najlepszym rozwiązaniem lekarza.
chociaż rzadko, warunek nie musi być nierozpoznany. Rozpoznając kluczowe cechy kliniczne, lekarze mogą ujawnić przyczynę objawów swojego pacjenta i zyskać pewność pełnego wyzdrowienia.
Dr Wang i Cantrell są optometrystami personelu w Orlando VA Medical Center. Dr Cali jest optometrystą w klinice Lee County VA. Autorzy pragną podziękować Josephowi Millerowi, od, Paulowi Gruosso, OD i Vanessie Santos, OD, za pomoc przy tym manuskrypcie.
1. Collier J. Peripheral neuritis: the Morisa lectures. Edinburgh Med J. 1932: 39: 601-18.
2. Fisher M. nietypowy wariant ostrego idiopatycznego zapalenia wielonerwowego (zespół ophthalmoplegia, ataksja i arefleksja). N Eng J Med. 1956:255:57-65.
3. Lo YL, et al. Spektrum kliniczne i immunologiczne zespołu Millera Fishera. Nerw Mięśniowy. 2007:36(5):615-27.
4. Mori m, Kuwabara S, Fukutake T, et al. Cechy kliniczne i rokowanie zespołu Millera Fishera. Neurologia. 2001:56(8): 1105-6.
5. Mori m, Kuwabara S, Fukutake T, et al. Plasmapharesis i zespół Millera Fishera: Analiza 50 kolejnych przypadków. J Neurol Neurochirurg Psych. 2002:72:680.
6. Yuan CL, Wang yj, Tsai CP. Miller Fisher syndrome: a hospital based retrospektywne badanie. Euro J Neurol. 2000:44(2):79-85.
7. Cheng BC, Chang WN, Chang CS et al. Zespół Guillain-Barré w południowym Tajwanie: cechy kliniczne, czynniki prognostyczne i wyniki terapeutyczne. Euro J Neurol. 2003:10 (6):655-62.
8. Berlit P, Rakicky J. zespół Millera Fishera. Przegląd literatury. J clinical neuro-ophthalmology. 1992:12:57-63.
9. Lyu RK, Tang LM, Cheng SY i in. Zespół Guillain-Barré na Tajwanie: badanie kliniczne z udziałem 167 pacjentów. J Neurol Neurochirurg Psych. 1997:63:494-500.
10. Tatsomoto m, Odaka m, Hirata K, Yuki N. izolowane porażenie uprowadzeń jako regionalna odmiana zespołu Guillain-Barré. J Neurol Sci. 2006:243:35-8.
11. Weiss ja, White JC. Korelacja przewodzenia aferentnego 1A z ataksją zespołu Fishera. Nerw Mięśniowy. 1986:9:327-32.
12. Das P, Biswash s, et al. Miller Fisher variant of guillain-barré syndrome: a case report and clinical review. Bangabandhu Sheikh Mujib Medical Univer J. 2012:5(1): 69-71
13. Kornberg AJ, Pestronk a, Blume GM, et al. Selektywne barwienie warstwy cząsteczkowej móżdżku przez IGG surowicy Millera Fishera i pokrewne zespoły. Neurologia. 1996:47:1317-20.
14. Inoue N, Koh C, Iwahashi T. wykrywanie surowicy przeciwciał antycerebellar u pacjentów z zespołem Millera Fishera. Euro J Neurol. 1999:42:230-4.
15. Barontini F, Di Lollo S, Maurri s, Lambruschini P. Localization of the pathological process in Miller Fisher Syndrome. Włochy J Neurol Sci. 1992:13:221-5.
16. Koga M, Yuki N, Takahashi m, et al. Bliskie powiązanie przeciwciał anty-gangliozydowych IgA z poprzedzającym je zakażeniem Campylobacter jejuni w zespołach Guillain-Barré i Fishera. J Neuroimmunol. 2003:141:112-7.
17. Guiloff RJ. Przewodnictwo nerwów obwodowych w zespole Millera Fishera. J Neurol Neurochirurg Psychiat. 1977:40:801-7.
18. Chikakiya H, Kunishige m, Yoshino h, et al. Opóźniona neuropatia ruchowa i czuciowa u pacjenta z zapaleniem mózgu pnia mózgu. J Neurol Sci. 2005:234:105-8.
19. Arakata Y, Yoshimuro m, Kobayashi s, et al. Zastosowanie dożylnej immunoglobuliny w zespole Millera Fishera. Brain Develop J. 1993:15:231-3.
20. Matsumoto H, Kobayashi O, Tamura K, et al. Zespół Millera Fishera ze śpiączką przejściową: porównanie z zapaleniem mózgu pnia mózgu Bickerstaffa. Brain Develop J. 2002:24:98-101.
21. Yeh JH, Chen WH, Chen JR, et al. Zespół Millera Fishera z centralnym zaangażowaniem: skuteczne leczenie plazmaferezą. Therap Aferesis Dializa J. 1999:3:69-71.
22. Papanikolaou T, Cath G, Boothman B, et al. Opis przypadku: ostra obustronna ophthalmopresis z źreniczkami saflexical mydriasis w zespole Millera-Fishera leczonych immunoglobuliną dożylną. J Ophthalmol. 2010; ID artykułu 291840.
23. Sandler RD, Hoggard N, Hadjivassiliou M. Miller-Fisher syndrome: czy ataksja jest centralna czy obwodowa? Ataksja Móżdżku. 2015:2:3.
24. Ejma m, Waliszewska-Prosół m, Hofman A, et al. Postępujący podostry zespół Millera-Fishera skutecznie leczony plazmaferezą. Polski J Neurol Neurochirurg. 2015;49(2):137-8.
25. San-Juan OD, Martínez-Herrera JF, Moreno García J, et al. Zespół Millera Fishera: 10 lat doświadczenia w ośrodku trzeciego poziomu. Euro Neurol. 2009; 62:149-54
26. Snyder LA, Rismondo V, Miller NR. Wariant Fisher zespołu Guillain-Barré (zespół Fishera). J Neurooftalmol. 2009 Dec;29(4):312-24.
27. Ito M, Kuwabara S, Odaka m, et al. Zapalenie mózgu pnia mózgu Bickerstaffa i zespół Fishera tworzą ciągłe spektrum: analiza kliniczna 581 przypadków. J Neurol. 2008;255:674-82.
28. Yuki N. ostre niedowłady mięśni zewnątrzgałkowych związane z przeciwciałem IgG anty-GQ1b. Ann Neurol 1996; 39: 668-72.