Konstrukcja sond FRET-TaqMan do multipleksowego PCR w czasie rzeczywistym z wykorzystaniem wewnętrznej kontroli pozytywnej
wprowadzenie
Real-time PCR umożliwia ciągłe monitorowanie fluorescencji fluoroforów podczas generowania produktów PCR w zamkniętym formacie rurki. Obecnie dostępne metody wykorzystują znakowane sondy lub barwnik interkalujący DNA do monitorowania amplifikacji produktu PCR. Sonda TaqMan jest jednoniciowym oligonukleotydem zawierającym fluorofor i gaszenie umieszczone 10-30 zasad od siebie. Sonda TaqMan jest hydrolizowana podczas amplifikacji z powodu 5′ -3 ’ specyficznej eksonukleazy dwuniciowej polimerazy Taq, która oddziela fluorofor od gaszenia, powodując wzrost fluorescencji. Przyrządy PCR w czasie rzeczywistym są wyposażone w detektory fluorescencji i oprogramowanie zdolne do szacowania progu cyklu (ct), cyklu, w którym fluorescencja jest większa niż fluorescencja tła, dla pozytywnych reakcji. Jednakże, technika nie może odróżnić prawdziwego ujemnego wyniku od fałszywie ujemnego, gdy inhibitory amplifikacji wpływają na PCR. Po ekstrakcji kwasem nukleinowym donosi się, że inhibitory mogą nadal być obecne w próbkach klinicznych (np. hemoglobina), próbkach środowiskowych (np. kwasy humusowe i fulwowe) i chemikaliach stosowanych podczas ekstrakcji kwasem nukleinowym (np. Etanol, detergenty lub środki chaotropowe). Wiarygodność testów diagnostycznych zwiększa się poprzez włączenie kwasu nukleinowego kontroli wewnętrznej, który może wskazywać na obecność i wpływ inhibitorów PCR (1,2). Wewnętrzna kontrola pozytywna (IPC) jest wzmacniana jednocześnie w obecności sekwencji docelowej przy użyciu znakowanego fluoroforu, który emituje światło o innej długości fali niż fluorofor używany do oznaczania sekwencji docelowej, przy czym dwa fluorofory wykrywane są w różnych kanałach przez instrument PCR w czasie rzeczywistym. Powszechnie stosowaną kontrolą wewnętrzną dla PCR jest plazmid zawierający sekwencję podobną do sekwencji docelowej z wyjątkiem obszaru sondy. Ograniczona liczba cząsteczek IPC jest dodawana do indywidualnego celu testu i co-amplifikowana z docelowym kwasem nukleinowym; tak więc dodatni sygnał IPC jest dowodem na to, że reakcja amplifikacji przebiegała wystarczająco długo, aby wygenerować pozytywny sygnał z bardzo małych ilości docelowego kwasu nukleinowego. Ta cecha jest ważna dla zapewnienia równoważnej amplifikacji IPC i docelowego kwasu nukleinowego (3-9). Jednak niektóre urządzenia—takie jak LightCycler 1.2 i LightCycler 2 (Roche Applied Science, Indianapolis, IN, USA), Ruggedized Advanced Pathogen Identification Device (R. A. P. I. D.) instrument (Idaho Technology, Salt Lake City, UT, USA) i ręczne Instrumenty PCR w czasie rzeczywistym-są wyposażone tylko w jedno źródło światła i powiązany kanał emisyjny. Multipleksowanie na tego typu instrumentach wymaga użycia multi-znakowanych oligonukleotydów i koncepcji fluorescencyjnego rezonansowego transferu energii (FRET). Jednak, gryźć sondy mogą być trudne do projektowania i może brak solidności w regionach hiperwariable i gdy nieoptymalne warunki amplifikacji są obecne. Zastosowanie sond progu wymaga identyfikacji dwóch regionów dla dwóch sond. System probe FRET opiera się na sąsiedniej hybrydyzacji oligonukleotydów dwóch pojedynczych znakowanych sond w taki sposób, że fluorescencja barwnika akceptora jest wykrywana przez wzbudzenie fluoroforu dołączonego do drugiej sondy. Główną wadą sondy FRET jest wymóg większego obszaru sekwencji niezbędnego do umieszczenia dwóch sąsiednich sond z przerwą 2-5 nukleotydów. Istnieje możliwość multipleksowania z innymi systemami etykiet, takimi jak MegaStokes dye (DYXL; Dyomics, Jena, Niemcy) i Pulsar 650 (Biosearch Technologies, Inc., Novato, CA, USA), ale te systemy etykietowania wymagają informacji spektralnych w celu skompensowania przesłuchu kanału i nie są powszechnie dostępne. Opracowana w tym badaniu sonda FRET-TaqMan nie wymaga jednak oprogramowania do kompensacji kolorów i wykorzystuje fluorofory (Fam, Cy5. 5), które są powszechnie dostępne. Niniejszy raport koncentruje się na konstrukcji potrójnie znakowanej sondy umożliwiającej multipleksowanie w instrumentach wyposażonych tylko w jedno źródło wzbudzenia z niebieską diodą elektroluminescencyjną (LED) (470 nm) i powiązane trzy odpowiednie kanały detekcji (np. 530 nm, 640 nm i 705 nm).
materiały i metody
zasada projektowania sondy
wszystkie eksperymenty przeprowadzono na systemie R. A. P. I. D. Idaho Technology, przenośnym przyrządzie PCR w czasie rzeczywistym z 32 próbkami, wyposażonym w niebieską diodę LED do wzbudzenia (470 nm) i powiązane trzy kanały detekcji fluorescencji, które mierzą fluorescencję przy 530, 640 i 705 nm dla testów mono-i dwukolorowych. Startery jvaf i JVAR (10) oraz sonda 5′-FAM/BHQ-1 zostały zsyntetyzowane w CDC Biotechnology Core Facility (Atlanta, GA, USA), a sonda 5 'FAM-Cy5.5/3′ BHQ-3 została zsyntetyzowana w Idaho Technology. Aby umożliwić duplex TaqMan PCR, jedna sonda została oznaczona 5′-FAM/BHQ-1 (długość fali fluorescencji 520 nm), a druga 5 'FAM-Cy5, 5/3′ BHQ-3 (długość fali fluorescencji 705 nm). W roztworze sonda oznaczona 5 'FAM-Cy5, 5/3′ HQ-3 pozostaje hartowana po hybrydyzacji z sekwencją, a po kolejnym rozszczepieniu sondy przez enzymatyczne przedłużenie startera, gaszenie jest eliminowane. Powoduje to FRET poprzez wzbudzenie FAM przez źródło światła instrumentu R. A. P. I. D. Energia wzbudzenia jest przenoszona do akceptora fluoroforu, Cy5.5, a emitowana fluorescencja jest mierzona w drugim kanale fluorescencyjnym (kanał fluorescencyjny F3 dla instrumentu R. A. P. I. D.). Emisja fluorescencji Cy5. 5 jest wynikiem połączenia technik sondy FRET i TaqMan, więc te potrójnie znakowane sondy mogą być określane jako sondy „FRET-TaqMan”.
startery, sondy i plazmid
aby zademonstrować możliwości multipleksowania projektu FRET-TaqMan, użyliśmy specyficznego dla rodzaju testu PCR Cryptosporidium w czasie rzeczywistym do amplifikacji części genu 18s rRNA o długości 159 bp (10). Bezpośredni podkład (JVAF) był 5′-ATGACGGGTAACGGGGGAAT-3′, zwrotny primer (JVAR) był 5′-CCAATTACAAAAC-CAAAAAGTCC-3′, a sonda TaqMan (JVAP) był 5′-FAM-CGCGCCTGCT-GCCTTCCTTAGATG-BHQ1–3′, co odpowiada нуклеотидам 100-118, 258-236 i 184-161, odpowiednio, numer rejestracyjny GenBank. AY458612 Startery do budowy Cryptosporidium IPC zawierały ten sam zestaw sekwencji starterowych zgłoszonych do amplifikacji Cryptosporidium spp., z wyjątkiem obszaru wiążącego sondę docelowego DNA, który został zastąpiony Sztuczną sekwencją 5 ’- TTGTGCTCGTAGTCGCCTCTCTGTCTCT-3 ’ (JPIPC). Ta sekwencja sondy miała temperaturę wyżarzania 69°C i została zaprojektowana tak, aby nie miała struktury wtórnej. Teoretyczna specyficzność dla wewnętrznej sondy została ustalona przez badanie wybuchowe (11) sondy i sekwencji starterowych, w których nie znaleziono dokładnego dopasowania do żadnej sekwencji Cryptosporidium. Fragment IPC zsyntetyzowano i sklonowano do plazmidu za pomocą zintegrowanych technologii DNA (Coralville, IA, USA). Znane stężenie plazmidowego DNA (100 kopii/reakcja) dodano do mieszanki wzorcowej wszystkich reakcji z wyjątkiem kontroli negatywnej. Potrójnie znakowana sonda (jpipc) specyficzna dla IPC została zsyntetyzowana za pomocą technologii Idaho, jak pokazano na rysunku 1. Sondę oligo-nukleotydową FAM / Cy5. 5 / BHQ3 zsyntetyzowano zaczynając od końca 3′ za pomocą Nie fluorescencyjnego szkła kontrolowanego porów Czarnej Dziury (BHQ-3) (CPG). Następnie 5 'koniec został sprzężony z fosporamidytem Cy5, 5, a następnie z końcowym 5′ fluoresceiną (6-FAM) fosporamidytem.
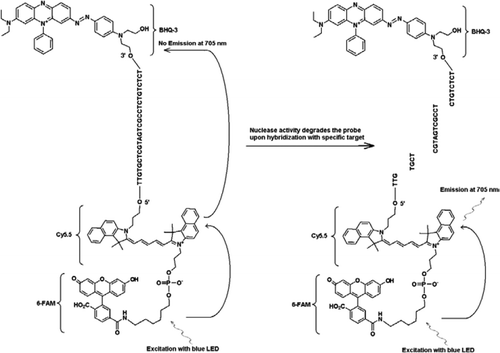
sonda FRET-TaqMan zawiera trzy etykiety: grupę tłumiącą czarną dziurę na końcu ramienia 3′, emiter fluoroforu (Cy5.5) i kombajn fluoroforu (FAM) połączone ze sobą na końcu ramienia 5′. FAM skutecznie absorbuje energię z niebieskiej diody LED jako źródła światła. W przypadku braku celów sonda jest ciemna, ponieważ energia pochłaniana przez FAM jest przenoszona do Cy5. 5, a z kolei energia uwalniana z Cy5.5 jest przenoszony do hartownika i tracony jako energia cieplna. W obecności celów, sonda TaqMan jest rozszczepiana przez aktywność polimerazy Taq, a hartownik jest uwalniany, a energia absorbowana przez FAM jest przenoszona do Cy5.5 przez mechanizm progu, aby emitować fluorescencję przy 705 nm.
test PCR w czasie rzeczywistym
cytometria przepływowa posortowane oocysty Cryptosporidium parvum (200 oocyst na probówkę) uzyskane z University of Wisconsin ’ s State Laboratory of Hygiene (Madison, WI, USA). DNA z C. oocysty parvum ekstrahowano za pomocą protokołu opisanego przez Hilla i wsp. (12) i zawieszone w 80 µL buforu Tris EDTA (TE, pH 8,0). Na reakcję dodano dwa mikro-litry DNA, co odpowiadało 5 oocystom (co odpowiada 100 kopiom docelowego DNA). Mieszanina reakcyjna amplifikacji składała się z mieszaniny reakcyjnej Quantifast Probe PCR + ROX kit (Cat. nr 204354; Qiagen) bez dodatku ROX, jvaf, startery JVAR (0,4 µM każdy), sonda JVAP (0,2 µM), sonda JPIPC (0,2 µM) i 100 kopii IPC. Do kapilary reakcyjnej (Roche Diagnostics, Indianapolis, IN, USA) dodano porcję (2 µL) wyekstrahowanej próbki DNA zawierającą 18 µL mieszaniny reakcyjnej. Dodatnia Kontrola DNA genomowego C. parvum (fig.2, fig. 1), plazmid IPC (fig. 2, fig. 2), mieszanina kontroli dodatniej C. parvum i IPC (fig. 2, fig. 3) oraz kontrola bez szablonu (NTC) składająca się z wody wolnej od nukleaz (Cat. nie. Am9932; Ambion, Foster City, CA, USA) zamiast DNA (Rysunek 2, Wykres 4) zostały uwzględnione w każdym eksperymencie. Wykonanie protokołu z następującymi warunkami PCR zajęło ∼30 minut: etap denaturacji na gorąco w temperaturze 95 ° C przez 3 minuty, następnie 45 cykli z denaturacją w temperaturze 95°C przez 5 s, wyżarzanie w temperaturze 60°C przez 30 s z pojedynczym pozyskiwaniem fluorescencji (z szybkością rampy temperatury 20°C/s). Dodatni wynik odnotowano dla FAM w kanale F1 (530 nm) i dla Cy5, 5 w kanale F3 (705 nm). Reakcje amplifikacji przeprowadzono w trzech egzemplarzach. Amplikony (159 bp) wizualizowano za pomocą elektroforezy w żelu agarozowym i Jaskrawozielonego barwienia kwasem nukleinowym (eenzym, Gaithersburg, MD, USA) w celu potwierdzenia specyficzności produktu PCR.
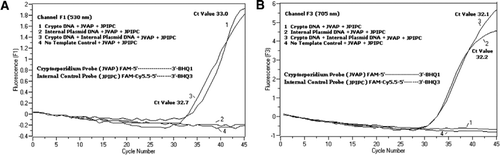
we wszystkich mieszaninach reakcji PCR dodano równomolarną mieszaninę FAM/BHQ1 i Fam/Cy5, 5/BHQ3. A) kanał F1 monitoruje wzrost fluorescencji FAM na obecność C. parvum. Wykres 1 (kanał F1 głównie zbiera sygnał FAM) zawierający tylko DNA C. parvum ujawnił w PCR specyficzny dla Kryptosporidium sygnał FAM. Wykres 2 zawierał tylko plazmidowe DNA i brak sygnału FAM z sondy Fam/Cy5.5 / BHQ3. Działka 3 zawierała zarówno C. parvum i plazmid DNA, ale wytwarzały tylko sygnał FAM specyficzny dla C. parvum. Wykres 4 był ujemną próbką zawierającą tylko wodę wolną od nukleaz zamiast jakiegokolwiek wzorca DNA; nie zaobserwowano fluorescencji z sond. B) kanał F3 monitoruje wzrost Cy5. 5 pod kątem obecności plazmidu kontroli wewnętrznej. Wykres 1 zawierał tylko DNA C. parvum i brak sygnału fluorescencyjnego z sondy FAM / BHQ1. Wykres 2 (kanał F3 głównie zbiera sygnał Cy5.5) zawierający tylko plazmid DNA ujawnił wewnętrzny sygnał Cy5.5 specyficzny dla kontroli plazmidów (IPC) w PCR. Działka 3 zawierała zarówno C. parvum i plazmid DNA, ale wytwarzały tylko sygnał Cy5.5 specyficzny dla IPC. Wykres 4 był ujemną próbką zawierającą tylko wodę wolną od nukleaz w miejsce jakiegokolwiek wzorca DNA; nie zaobserwowano fluorescencji z sond.
wyniki i dyskusja
sygnały fluorescencyjne PCR w czasie rzeczywistym odzwierciedlające amplifikację genomowego DNA C. parvum wykryto w kanale F1 (Fig. Sygnał fluorescencyjny odzwierciedlający wzmocnienie IPC wykryto w kanale F3 (Fig.2b) z powodu FAM/Cy5.5 znakowanie sondy IPC TaqMan. Sonda ta wywołała wzrost liczby Cy5.5 w kanale F3 (Cy5.5), ale nie wzrost liczby FAM w kanale F1 (FAM), gdy BHQ3 jest oddzielany od FAM/Cy5.5 z powodu aktywności nukleazy, ponieważ energia została przeniesiona z FAM do Cy5.5. Podobnie, w reakcji dupleksu, amplifikację genomowego DNA C. parvum wykryto w kanale F1, podczas gdy amplifikację IPC wykryto jednocześnie w kanale F3. FAM ma wzbudzenie przy 495 nm i emisję przy 520 nm. Cy5. 5 ma wzbudzenie 690 nm i emisję 705 nm. Przy tak bliskiej odległości między Cy5. 5 i FAM na potrójnie znakowanej sondzie IPC, fluorofor Cy5.5 emituje fluorescencję z powodu całkowitego przeniesienia energii z fluoroforu FAM, co powoduje brak emisji fluorescencji FAM w kanale F1 (Fig. 2A). Negatywne reakcje kontrolne nie wykazały wzrostu fluorescencji z powodu FAM lub Cy5. 5. Wyniki te pokazują również, że pojedyncze niebieskie (470 nm) źródło LED jest wystarczające do monitorowania podwójnej emisji fluorescencji przez kanały F1 (530 nm) i F3 (705 nm) bez nakładania się spektralnego w odpowiednich kanałach monitorowania dla FAM i Cy5.5. Technikę FRET-TaqMan przetestowano również przy użyciu panelu 12 ekstraktów DNA z próbek kału (8 z próbek wcześniej oznaczonych jako pozytywne dla Cryptosporidium spp. i cztery okazy, które nie wykazywały obecności Cryptosporidium spp.). Test 18s rRNA był w stanie wykryć wszystkie osiem Cryptosporidium spp. Stwierdzono, że test IPC TaqMan wzmacnia tylko DNA plazmidowe IPC. W teście IPC uzyskano wartości Ct 32-37, które wskazywały do 5 wartości CT hamowania PCR dla niektórych próbek kału (32 była oczekiwaną wartością ct dla reakcji bez hamowania).
niniejsza technika różni się od tradycyjnie stosowanego progu tym, że stosuje się jedną fluorescencyjnie znakowaną sondę oligonukleotydową (sondę FRET-TaqMan) zamiast dwóch sond (sondy FRET), w których jedna sonda zawiera fluorofor dawcy, a druga sonda zawiera fluorofor akceptora (13,14). Probe FRET – TaqMan jest oznaczony na 5 'koniec z dwoma fluorophores dawcy (FAM) i akceptor (Cy5.5) fluorophores i na 3′ Koniec z gaszenia BHQ-3 jak sonda TaqMan. Unikalny obszar wiązania sondy jest wybrany w celu odróżnienia od docelowego DNA, tak aby monitorować dwa różne pomiary fluorescencji w dwóch różnych kanałach detekcji (530 nm W F1 i 705 nm W F3) z tej samej reakcji amplifikacji bez potrzeby dołączania oprogramowania do kompensacji kolorów. Docelowe DNA i IPC miały te same miejsca wiązania startera, ale inną sekwencję sondy. Zapewnia to, że użycie tych samych starterów dla obu celów DNA w celu zminimalizowania powstawania primer-dimer lub interakcji między zestawami starterów. Gdy fluorofory Fam / Cy5. 5 są umieszczone blisko końca 5′, transfer energii rezonansowej odbywa się wraz ze wzrostem emisji fluorescencji Fluoroforu Cy5.5. Jednakże zastosowanie BHQ3, nie fluorescencyjnego akceptora dla Cy5.5, zmniejsza potencjalną fluorescencję tła, co skutkuje wyższym stosunkiem sygnału do szumu. Stwierdzono, że dodanie 100 kopii IPC do reakcji zawierającej ∼100 kopii genomu C. parvum DNA dało podobne wartości Ct (CT = 32), gdy test Cryptosporidium TaqMan był wykonywany jako test singleplex i jako test duplex z testem IPC TaqMan. Po nałożeniu na panel próbek DNA z dodatnim Cryptosporidium, IPC wskazało, że niektóre próbki nie zawierają inhibitorów PCR (na podstawie oczekiwanych wartości Ct 32), ale inne próbki były związane z hamowaniem do 5 wartości Ct.
nowo opracowana wewnętrzna kontrola dodatnia PCR okazała się wrażliwa i specyficzna dla jednoczesnego wykrywania dwóch celów w tej samej reakcji dzięki oznakowaniu FAM/Cy5.5-BHQ3 i FAM / BHQ1 dla odpowiedniego genu docelowego IPC i 18S rRNA w dwukolorowym multipleksie PCR do monitorowania w kanałach fluorescencyjnych F1 i F3 instrumentu R. A. P. I. D. Zastosowanie sondy oznaczonej symbolem 5 'FAM-Cy5.5/3′ BHQ-3 jako drugiego reportera jest korzystne, ponieważ maksimum emisji dwóch sond były dobrze oddalone od siebie. Konstrukcja potrójnie znakowanej sondy FRET-TaqMan może mieć zastosowanie w opracowywaniu niedrogiego, jednorazowego urządzenia „lab-on-a-chip” z optyką składającą się z jednego niebieskiego wzbudzenia LED i dwóch źródeł emisji 530 nm i 705 nm, z odpowiednimi filtrami optycznymi do wykrywania FAM w jednym kanale i Cy5.5 w drugim. Połączenie to nie wymagałoby dwóch wysoko zasilanych źródeł fluorescencji i systemów detekcji.
podziękowania
autorzy doceniają pomoc Mike ’ a Powersa w Zakładzie sekwencjonowania DNA i rdzenia genomowego (University of Utah, Salt Lake City, UT, USA) oraz Rachelle Muller w Idaho Technology, Inc. do dyskusji na temat rozwoju wewnętrznej kontroli pozytywnej dla cyklera R. A. P. I. D. Publikacja ta była częściowo wspierana przez fundusze udostępnione za pośrednictwem Centers for Disease Control and Prevention, biura koordynującego gotowość na terroryzm i reagowanie w sytuacjach kryzysowych. Używanie nazw handlowych i komercyjnych źródeł służy wyłącznie identyfikacji i nie oznacza poparcia przez Centers for Disease Control and Prevention lub Departament Zdrowia i usług ludzkich Stanów Zjednoczonych.
- 1. Hartman, L. J., S. R. Coyne, and D. A. Norwood. 2005. Opracowanie nowej wewnętrznej kontroli pozytywnej dla testów opartych na Taqman. Mol. Cell. Sondy 19:51-59.Crossref, Medline, CAS, Google Scholar
- 2. Hoorfar, J., B. Malorny, A. Abdulmawjood, N. Cook, M. Wagner, and p. Fach. 2004. Praktyczne rozważania przy projektowaniu wewnętrznych sterowników wzmocnienia do diagnostycznych testów PCR. J. Clin. Mikrobiol. 42:1863–1868.Crossref, Medline, CAS, Google Scholar
- 3. Betsou, F., K. Beaumont, J. M. Sueur, and J. Orfila. 2003. Budowa i ocena DNA kontroli wewnętrznej do amplifikacji DNA Chlamydia trachomatis metodą PCR z próbek moczu. J. Clin. Mikrobiol. 41:1274–1276.Crossref, Medline, CAS, Google Scholar
- 4. Burggraf, S. and B. Olgemoller. 2004. Prosta technika wewnętrznej kontroli testów wzmocnienia w czasie rzeczywistym. Clin. Chem. 50:819–825.Crossref, Medline, CAS, Google Scholar
- 5. Drosten, C., M. Weber, E. Seifried, and W. K. Roth. 2000. Ocena nowego testu PCR z konkurencyjną sekwencją kontroli wewnętrznej dla badań przesiewowych dawców krwi. Transfuzja 40: 718-724.Crossref, Medline, CAS, Google Scholar
- 6. Gruber, F., F. G. Falkner, F. Dorner, and T. Hammerle. 2001. Ilościowy wirusowego DNA przez real-time PCR stosując amplifikację dupleksu, wewnętrzną standaryzację i dwukolorową detekcję fluorescencji. Appl. Environ. Mikrobiol. 67:2837–2839.Crossref, Medline, CAS, Google Scholar
- 7. Rosenstraus, M., Z. Wang, S. Y. Chang, D. DeBonville i J. P. Spadoro. 1998. Wewnętrzna kontrola rutynowej diagnostyki PCR: projekt, właściwości i wpływ na wydajność kliniczną. J. Clin. Mikrobiol. 36:191–197.Medline, CAS, Google Scholar
- 8. Stocher, M., V. Leb, and J. Berg. 2003. Wygodne podejście do generowania wielu DNA kontroli wewnętrznej dla panelu testów PCR w czasie rzeczywistym. J. Virol. Metody 108:1-8.Crossref, Medline, CAS, Google Scholar
- 9. Zimmermann, K. and J. W. Mannhalter. 1996. Techniczne aspekty konkurencyjnej ilościowej PCR. BioTechniques 21:268-279.Link, CAS, Google Scholar
- 10. Jothikumar, N., A. J. da Silva, I. Moura, Y. Qvarnstrom, and V. R. Hill. 2008. Wykrywanie i różnicowanie Cryptosporidium hominis i Cryptosporidium parvum przez podwójne testy Taqmana. J. Med. Mikrobiol. 57:1099–1105.Crossref, Medline, CAS, Google Scholar
- 11. Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Podstawowe narzędzie do lokalnego wyszukiwania wyrównania. J. Mol. Biol. 215:403–410.Crossref, Medline, CAS, Google Scholar
- 12. Hill, V. R., A. M. Kahler, N. Jothikumar, T. B. Johnson, D. Hahn, and T. L. Cromeans. 2007. Wielostanowa ocena procedury opartej na ultrafiltracji do jednoczesnego odzyskiwania drobnoustrojów jelitowych w 100-litrowych próbkach wody z kranu. Appl. Environ. Mikrobiol. 73:4218–4225.Crossref, Medline, CAS, Google Scholar
- 13. Bernard, P. S., M. J. Lay, and C. T. Wittwer. 1998. Zintegrowana amplifikacja i wykrywanie mutacji punktowej c677t w genie reduktazy metylenotetrahydrofolianowej za pomocą rezonansowego transferu energii fluorescencji i krzywych topnienia sondy. Anal. Biochem. 255:101–107.Crossref, Medline, CAS, Google Scholar
- 14. Wittwer, C. T., M. G. Herrmann, A. A. Moss, and R. P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130–138.Link, CAS, Google Scholar