Debole nelle ginocchia: Sindrome di Miller Fisher
La sindrome di Miller Fisher (MFS), una rara variante autolimitante della sindrome di Guillain-Barré (GBS), è una sindrome da anticorpi anti-GQ1bIgG che colpisce il sistema nervoso periferico e centrale.1 Una condizione neuromuscolare polineuropatica acuta, MFS provoca una paralisi ascendente con una classica triade di presentazione di oftalmoplegia, atassia e areflessia. I ricercatori hanno descritto per la prima volta la triade clinica nel 1932.1 Due decenni dopo, lo specialista canadese di ictus Charles Miller Fisher fu il primo a pubblicare un rapporto in 1956 che descrisse i risultati clinici e definì la condizione come una forma limitata di GBS.2
I sintomi si verificano per diversi giorni, spesso in inverno e in primavera, in genere a seguito di un’infezione virale, e i pazienti spesso sperimentano la diplopia come uno dei primi sintomi. Quando un paziente presenta sintomi come esotropia intermittente, attività orizzontali compromesse e pupille midriatiche fisse, e il tuo esame neurologico è anormale, è tempo di pensare a GBS, in particolare MFS. Il caso seguente illustra i segni e i sintomi rilevanti e riflette il risultato tipico.
Storia
Un maschio caucasico di 27 anni si è presentato alla clinica oculistica VA durante i mesi primaverili riportando diplopia binoculare ad esordio acuto iniziata un giorno prima. Il paziente ha dichiarato che era costante, ma si è verificato solo a distanza di visualizzazione con oggetti spostati orizzontalmente. Non c’era dolore agli occhi o visione offuscata associata alla diplopia. Si è anche lamentato delle recenti vertigini e difficoltà di equilibrio. Ha riportato un’infezione del seno con una significativa congestione paranasale iniziata una settimana prima della visione doppia. Precedenti storie oculari e mediche erano insignificanti; il paziente non stava attualmente assumendo alcun farmaco e non aveva allergie ai farmaci.
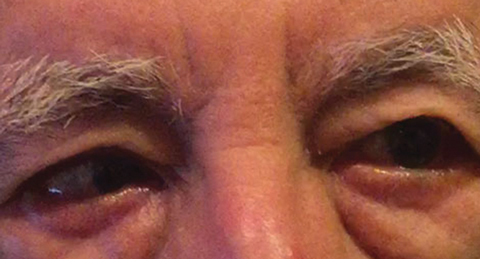
Fig. 1. La diagnosi differenziale dopo l’esame basale includeva una paralisi parziale della CN VI destra, che potrebbe indicare una condizione vascolare, come visto qui in un altro paziente. Clicca sull’immagine per ingrandirla. Foto: Michael DelGiodice, OD
Dati diagnostici
All’esame basale, le acuità visive non corrette erano 20/20-2 OD e 20/25-2 OS. Le pupille erano di 5 mm ugualmente rotonde e reattive alla luce senza difetti pupillari afferenti. Le motilità extraoculari erano piene, lisce e precise in entrambi gli occhi senza restrizioni. Il test di copertura a distanza ha rivelato un’intermittente esotropia destra da quattro a sei prismi nello sguardo primario che si verifica circa il 50% delle volte. Esecuzione di test di copertura a quasi rivelato un quattro prisma diottrica esoforia in sguardo primario senza tropia.
Pur non intrapresa in questo caso, i medici dovrebbero coprire test in primario, sguardo destro e sinistro. Nei pazienti con diplopia, la paralisi del nervo cranico sei (CN VI) si manifesterà come deviazione non comitant, peggiore nello sguardo destro o sinistro, a seconda del lato in cui si trova la paralisi. Una phoria scompensante, d’altra parte, mostrerà una deviazione comitant.
L’errore di rifrazione era -0.50 D sphere OD e -0.75 D sphere OS e la visione era correggibile a 20/20 in ciascun occhio. Le pressioni intraoculari erano entro i limiti normali in entrambi gli occhi. I risultati del segmento anteriore e posteriore erano insignificanti negli occhi destro e sinistro. I nervi ottici apparivano sani con un rapporto tazza-disco di 0,15 in entrambi gli occhi.
Le diagnosi differenziali includevano una paralisi parziale del CN VI destro (Figura 1), miastenia grave, un processo compressivo e un’esoforia a distanza scompensante che causava un’esotropia destra intermittente. Il paziente ha negato qualsiasi precedente storia di diplopia binoculare.
L’imaging del cervello e delle orbite è stato ordinato dalla clinica oculistica per escludere il coinvolgimento cerebrale, inclusa una lesione compressiva. La tomografia computerizzata (CT) scansioni del cervello e delle orbite sono state eseguite e interpretate dal neuroradiologo dello staff. La TC è stata scelta al posto della risonanza magnetica (MRI) a causa della tempistica e della disponibilità; test e risultati potrebbero essere ottenuti lo stesso giorno con la TC in clinica dopo ore. I risultati sono stati insignificanti senza segni di emorragia intracranica, lesione compressiva o infarto acuto.
La TC dei seni paranasali ha rivelato una malattia del seno paranasale cronica da moderata a grave e una completa opacizzazione del seno mascellare destro. Non c’era alcuna estensione nell’orbita. Il medico di famiglia del paziente è stato informato dei sintomi del paziente, dei risultati degli esami oculistici e dei risultati dell’imaging TC e ha iniziato il trattamento per la sinusite mascellare con una dose orale di azitromicina di cinque giorni e ha istruito il paziente a seguire la clinica oculistica.
Il paziente è tornato cinque giorni dopo aver completato il suo corso di azitromicina orale e ha dichiarato di non aver sperimentato alcun miglioramento nella diplopia a distanza; inoltre, ha sperimentato l’insorgenza di fotofobia significativa e dolore perioculare intorno ad entrambi gli occhi. Il paziente ha dichiarato che anche il suo squilibrio dell’andatura e le difficoltà di deambulazione stavano peggiorando. All’esame, le acuità visive meglio corrette sono rimaste 20/20 negli occhi destro e sinistro. Le pupille sono state dilatate a 8,5 mm senza alcuna reattività alla luce in entrambi gli occhi (Figura 2). Le motilità extraoculari hanno rivelato una restrizione laterale con dolore al movimento nell’occhio destro e una restrizione mediale con dolore al movimento nell’occhio sinistro coerente con una paralisi dello sguardo destro (Figura 3). Gli inseguimenti orizzontali e verticali erano saccadici con movimenti imprecisi e a scatti. I risultati del segmento anteriore e posteriore sono rimasti insignificanti OU.
Le diagnosi differenziali dopo l’esame oftalmologico includevano oftalmoplegia internucleare sinistra, paralisi dello sguardo destro e paralisi dello sguardo destro con oftalmoplegia internucleare sinistra, che potrebbe spiegare i risultati della motilità extraoculare. Altri differenziali includono una lesione del mesencefalo o miastenia grave. Le diagnosi differenziali per la dilatazione della pupilla includono paralisi del terzo nervo (CN III), trauma e dilatazione farmacologica. Il paziente non ha avuto altri risultati coerenti con una paralisi del terzo nervo e ha negato traumi o esposizione ad agenti farmacologici.
Il paziente è stato sottoposto a neurologia per ulteriori valutazioni. L’esame neurologico ha riferito che il paziente era completamente vigile e orientato senza perdita di sensazione facciale. Il paziente ha mostrato una lieve atassia con difficoltà a camminare in tandem e in piedi da una posizione accovacciata. Erano presenti debolezza prossimale sia degli arti superiori che inferiori e riflessi anormali. Tutte le modalità sensoriali erano intatte. La risonanza magnetica e l’angiografia a risonanza magnetica (MRA) del cervello e delle orbite erano insignificanti. La proteina del liquido cerebrospinale è stata elevata a 75 mg / dL (intervallo di riferimento: da 15 mg / dL a 45 mg / dL) senza altre anomalie. Il test dell’elettromiogramma era insignificante e non mostrava anomalie nella conduzione nervosa dalla colonna vertebrale ai piedi e alle mani.
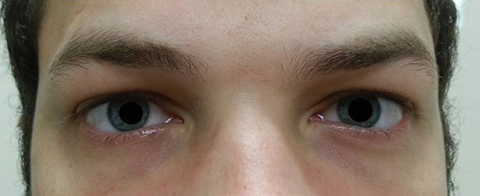
Fig. 2. Le pupille del paziente erano dilatate di 8,5 mm senza reattività negli occhi destro o sinistro. Clicca sull’immagine per ingrandirla.
Diagnosi
Il paziente presentava sintomi acuti di diplopia, atassia e areflessia. Data l’esotropia destra intermittente, attività orizzontali alterate, pupilla midriatica fissa, atassia lieve e riflessi anormali all’esame neurologico e proteine del liquido cerebrospinale elevate, è stata fatta la diagnosi MFS. Il paziente è stato ricoverato in ospedale per monitorare eventuali ulteriori complicazioni. Una benda sull’occhio è stata fornita al paziente per coprire l’occhio destro come necessario per la diplopia.
Non esistono attualmente criteri diagnostici pubblicati per la sindrome di Miller Fisher. La diagnosi è fatta solitamente con la presentazione della triade clinica con la rappresentazione e gli studi del liquido cerebrospinale. MRI è tipicamente insignificante nella condizione, anche se alcuni studi hanno mostrato lesioni centrali nel mesencefalo, pons e midollo inferiore. Studi elettrofisiologici mostrano una ridotta o anormale conduzione sensoriale periferica.17
La proteina del liquido cerebrospinale è spesso elevata senza altri risultati anormali. In uno studio storico, il 64,4% dei pazienti affetti da MFS ha mostrato un aumento della proteina del liquido spinale.8 Un test anticorpale IgG anti-GQ1b positivo consente una diagnosi definitiva. In uno studio, l’anticorpo anti-GQ1b IgG era presente fino al 95% dei pazienti con la condizione e assente nei controlli.3
Il neurologo ha diagnosticato il paziente con MFS basato sulla triade clinica di oftalmoplegia, atassia e areflessia, una risonanza magnetica insignificante e MRA del cervello e una proteina del liquido cerebrospinale elevata. Pertanto, il test degli anticorpi anti-GQ1b IgG non è stato eseguito in questo caso.
Diverse caratteristiche cliniche della MFS sono anche osservate nella sindrome di Guillain-Barré e nell’encefalite del tronco cerebrale di Bickerstaff, rendendo la diagnosi impegnativa. I pazienti con solo Guillain-Barré presentano debolezza degli arti, perdita sensoriale, neuropatia cranica e areflessia senza manifestazioni oftalmologiche. I pazienti con encefalite del tronco cerebrale di Bickerstaff tipicamente presenti con oftalmoplegia, atassia, iper-riflessione e una coscienza disturbata. Poiché parecchi sintomi e segni sono presenti in tutte e tre le circostanze, la prova del titolo dell’anticorpo di anti-GQ1b IgG è utile per una diagnosi più definitiva. L’anticorpo anti-GQ1b IgG è positivo in una grande maggioranza dei pazienti con sindrome di Miller Fisher rispetto al 66% nell’encefalite del tronco cerebrale di Bickerstaff e al 26% dei pazienti con sindrome di Guillain-Barré.16
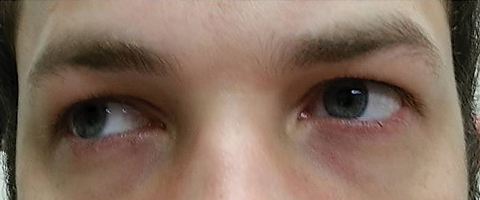
Fig. 3. Le motilità extraoculari hanno rivelato restrizioni nella destroversione. Clicca sull’immagine per ingrandirla.
Trattamento e follow-up
Il paziente è stato valutato il giorno dopo il ricovero, quando ha dichiarato di non aver riscontrato cambiamenti visivi o miglioramenti nell’equilibrio. La diplopia binoculare era ancora costante; pertanto, l’occhio destro rimaneva rattoppato. Dolore agli occhi con sguardi lontani e movimenti oculari veloci erano ancora presenti. In un esame limitato al capezzale, le acuità visive meglio corrette sono rimaste 20/20 negli occhi destro e sinistro. Le pupille erano dilatate a 8.0mm in entrambi gli occhi con una reattività minima, che è stata leggermente migliorata rispetto al giorno precedente. La visione a colori era normale negli occhi destro e sinistro. Le ostilità extraoculari erano piene in tutti i quadranti con dolore ancora presente su sguardi orizzontali lontani. Inseguimenti orizzontali sono rimasti saccadic con movimenti a scatti e nistagmo endpoint. I nervi ottici apparivano sani con margini distinti e un rapporto tazza-disco di 0,15 in entrambi gli occhi.
Due giorni dopo, il paziente ha dichiarato un miglioramento di tutti i sintomi e che i suoi occhi “si sentono meglio oggi di quanto non siano stati in un po’.”Il dolore oculare e la diplopia binoculare sono stati entrambi risolti. Le acuità visive sono rimaste 20/20 negli occhi destro e sinistro. Le dimensioni della pupilla erano 7,5 mm al buio e 7,0 mm alla luce con risposte dirette e consensuali migliorate in entrambi gli occhi. Le ostilità extraoculari erano piene in tutti i quadranti senza dolore agli occhi. Inseguimenti orizzontali sono rimasti saccadic con movimenti a scatti e nistagmo endpoint. L’esame neurologico ha rivelato riflessi anormali, che sono rimasti stabili. Le opzioni di trattamento per la MFS includono plasmaferesi e immunoglobuline IgG per via endovenosa (IV), ma queste sono state sospese dato il miglioramento dell’oftalmoplegia e dell’atassia.
Quattro giorni dopo, il paziente ha riferito che tutti i sintomi stavano ancora migliorando, con qualche difficoltà di messa a fuoco e tracciamento durante l’utilizzo del computer. L’equilibrio è stato risolto all ‘ 80% e ci sono stati pochi casi di diplopia a distanza. Acuities è rimasto 20/20 OU. Gli alunni erano 5,5 mm al buio e 4,0 mm alla luce con 3 + risposte dirette e consensuali OU. Le ostilità extraoculari erano piene in tutti i quadranti senza dolore agli occhi. Le attività orizzontali sono state migliorate, senza movimenti saccadici. Alloggio monoculare era normale, che è stato misurato 8,5 e 9 diottrie prisma OD e OS, rispettivamente.
Una settimana dopo, il paziente ha riferito che la sua visione è tornata quasi alla normalità senza diplopia e lievi difficoltà di messa a fuoco e tracciamento. Il saldo è stato risolto al 100%. Le acuità visive sono rimaste 20/20 negli occhi destro e sinistro. Le dimensioni della pupilla erano 5,5 mm al buio, 4,0 mm alla luce con 4 + risposte dirette e consensuali OU. Le ostilità extraoculari erano piene in tutti i quadranti senza dolore agli occhi. Le attività orizzontali sono migliorate senza movimenti saccadici osservati. La salute oculare era insignificante.
Il trattamento è stato sospeso data la risoluzione di oftalmoplegia e atassia con areflessia stabile. Il recupero completo di opthalmoplegia e atassia ha richiesto due settimane. La gestione ha incluso un attento monitoraggio e follow-up in quattro settimane; al paziente è stato consigliato di tornare prima se uno qualsiasi dei suoi sintomi è tornato.
Caratteristiche cliniche
In studi ben descritti di MFS, tutti i pazienti hanno presentato la triade clinica di oftalmoplegia, atassia e areflessia.4,5 L’insorgenza dei sintomi si verifica in genere nell’arco di diversi giorni e i pazienti subiscono un’infezione virale da una a quattro settimane prima della comparsa dei sintomi clinici.4,5 Secondo uno studio su 50 pazienti con la condizione, l’intervallo medio tra l’inizio dell’infezione e lo sviluppo dei sintomi neurologici è di otto giorni.4 Altri segni di MFS includono disturbi del linguaggio, difficoltà a deglutire e un’espressione facciale anormale con incapacità di sorridere o fischiare.
Caratteristiche oftalmologiche. I praticanti dovrebbero essere alla ricerca di midriasi, retrazione del coperchio, chiusura dell’angolo acuto e diplopia secondaria a paralisi nervose che interessano CN III, IV e VI.3,8,10,22,24 Diplopia è il sintomo oftalmologico più comune riportato in molti studi MFS.8,16,25,26,27 Diplopia è anche il sintomo iniziale riportato nel 38,6% dei pazienti 223 in uno studio di riferimento e nel 65% dei pazienti 466 in un secondo studio.8,27 Nel secondo studio, il 100% dei pazienti mostrava oftalmoplegia esterna, mentre l’oftalmoplegia interna era presente nel 35% dei pazienti.27 Oftalmoplegia esterna si riferisce alla compromissione dei muscoli extraoculari esterni. L’oftalmoplegia interna si riferisce alla compromissione dello sfintere pupillare e del muscolo ciliare. L’oftalmoplegia completa colpisce sia i muscoli esterni che quelli interni.
In uno studio retrospettivo su 19 pazienti con MFS, tutti presentati con una o più paralisi nervose che interessano i muscoli extraoculari.25 Altri studi mostrano che i pazienti con MFS possono manifestare paralisi multiple del nervo cranico, che possono manifestarsi bilateralmente.22,24,25 Una scoperta comune trovata fra i disordini positivi di IgG di anti-GQ1b è deficit di abduzione coerente con una paralisi unilaterale o bilaterale di CN VI.26,28 L’oftalmoplegia interna causa midriasi della pupilla, dove la costrizione della pupilla alla luce e/o vicino alla stimolazione può variare da minima a assente.8,26 In un rapporto, la midriasi era presente nel 42% dei pazienti. Il coinvolgimento della CN VII si è verificato in circa il 45,7% dei pazienti affetti da MFS causando debolezza facciale, incapacità di sorridere o fischiare.8
Atassia. La causa di questa caratteristica clinica in MFS non è completamente compresa. Esiste un dibattito sul fatto che l’atassia sia causata da una disfunzione centrale nel cervelletto o periferica. La prima ipotesi, proposta dal Dr. Fisher, postulava il coinvolgimento dei neuroni Ia-afferenti.2 Ciò è stato supportato da un secondo studio decenni più tardi, che ha mostrato anomalie delle fibre Ia-afferenti in MFS.11 È stato anche proposto un coinvolgimento anormale dei nervi periferici con una mancata corrispondenza tra l’input propriocettivo dei mandrini muscolari e le informazioni cinestetiche dei recettori articolari.12
Ci sono state anche prove a sostegno che l’atassia ha origine nel cervelletto. Una predominanza di anticorpi anti-GQ1b IgG nel cervelletto è stata trovata usando la colorazione immunocitochimica del cervelletto umano e ulteriormente confermata con studi western blot, che hanno mostrato un aumento dell’anticorpo anti-GQ1b IgG nei pazienti con MFS rispetto al controllo, sebbene il meccanismo per il coinvolgimento cerebellare non sia stato studiato.13,14 Nel 2015, i ricercatori hanno pubblicato un case report di un paziente MFS sottoposto a risonanza magnetica. C’è stato un rapporto N-acetilaspartato/creatina ridotto, suggerendo una disfunzione cerebellare, che è tornata alla normalità 2,5 mesi dopo il recupero.23
Areflessia. Questa caratteristica clinica, insieme alla depressione dei riflessi tendinei profondi, è un segno di coinvolgimento del sistema nervoso periferico nella MFS. Gli studi elettrofisiologici hanno convalidato la conduzione nervosa periferica anormale in GBS e MFS.3 In uno studio, l ‘ 81,6% dei casi di MFS ha presentato arefexia.8 Un altro studio di riferimento ha mostrato che tutti i pazienti presentavano areflessia, che era ancora presente sei mesi dopo l’insorgenza.4
Quanto è comune MFS?
I dati epidemiologici sulla sindrome di Miller Fisher sono limitati. L’incidenza di MFS è piuttosto rara, che si verifica in 0,09 ogni 100.000 persone all’anno.3 MFS si verifica più comunemente nei pazienti con sindrome di Guillain-Barré, dal 3% al 25%.4,6,7 Questa associazione si verifica più in pazienti di discendenza dell’Estremo Oriente, suggerendo una possibile componente genetica alla MFS. L’età media di insorgenza è 43, con una gamma di 13 a 78.5 MFS colpisce i maschi due volte tanto quanto le femmine con un rapporto di due a 1.03.5 MFS si verifica più nelle stagioni invernali e primaverili.5,6,8 La causa esatta della predilezione stagionale non è stata determinata, ma è probabilmente associata alla natura post-infettiva della circostanza poichè le infezioni batteriche e virali sono state trovate per innescare una risposta autoimmune che causa la produzione dell’anticorpo anti-GQ1b IgG in MFS.
Fisiopatologia
La fisiopatologia della sindrome di Miller Fisher non è ben compresa; tuttavia, esistono diverse ipotesi da studi immunologici e istologici. È ben noto che la circostanza si trova all’interno dello spettro delle sindromi dell’anticorpo di anti-GQ1b IgG con la sindrome di Guillain-Barré e l’encefalite del tronco cerebrale di Bickerstaff.
Il ganglioside GQ1b è un gruppo di lipidi complessi coinvolti nel sistema nervoso centrale e periferico. GQ1b è un componente della struttura della membrana plasmatica nei nervi cranici che forniscono i muscoli extraoculari ed è coinvolto nella funzione cellulare alla giunzione neuromuscolare presinaptica.
Un meccanismo autoimmune derivante da una precedente infezione scatenante produrrà l’anticorpo IgG anti-GQ1b, che danneggia la funzione del ganglioside GQ1b, causando la demielinizzazione. Studi istologici hanno mostrato demielinizzazione e gonfiore assonale nei nervi cranici periferici e oculomotori.L’anticorpo anti-GQ1bIgG è assente nei pazienti sani, ma positivo in oltre il 90% dei pazienti con MFS.16,25
Infezioni batteriche e virali sono state trovate per innescare una risposta autoimmune, causando la produzione dell’anticorpo anti-GQ 1b IgG. I seguenti agenti infettivi che sono stati associati a MFS includono polmonite da micoplasma, HIV, Campylobacter jejuni, influenza emofilica, Helicobacter pylori e virus di Epstein-Barr.4,16 In un rapporto che ha esaminato i pazienti 466 MFS, 90% ha avuto una malattia antecedente, inclusa infezione delle vie respiratorie superiori, diarrea o entrambi.27
Prognosi e trattamento
La sindrome di Miller Fisher è una malattia autolimitante e ha una prognosi complessivamente positiva. I sintomi generalmente migliorano dopo alcune settimane con un recupero completo che si verifica in genere in due o tre mesi. In uno studio, il recupero è iniziato a una mediana di 13 giorni dall’insorgenza dei sintomi e la completa risoluzione dell’oftalmoplegia e dell’atassia si è verificata in sei mesi.4,5 Recidive sono state riscontrate in meno del 3% dei casi.4,5
Sebbene la MFS sia tipicamente autolimitante, le complicazioni sistemiche sono state associate con la circostanza. Le infezioni respiratorie superiori sono state trovate in 56% – 76% dei pazienti di MFS, che possono progredire ad insufficienza respiratoria che richiede la ventilazione meccanica.4,18,19 Altre rare complicanze gravi includono cardiomiopatia, acidosi lattica e coma.3,20
Le opzioni di trattamento per MFS includono plasmaferesi, immunoglobuline IV IgG e monitoraggio senza trattamento. I ricercatori postulano che la plasmaferesi e le IgG IV possono essere efficaci per accelerare il tempo di risoluzione poiché l’anticorpo Gq1b è IgG in classe e l’emivita delle IgG è di circa 21 giorni, che è più lunga delle emivite da cinque a sei giorni di IgM e IgA.19 Tuttavia, non sono stati condotti studi randomizzati in doppio cieco controllati con placebo che indagassero questi trattamenti.
Uno studio retrospettivo non è riuscito a mostrare che la plasmaferesi ha modificato la possibilità di recupero completo e il tempo di risoluzione dell’atassia e dell’oftalmoplegia; inoltre, l’intervallo dall’inizio all’inizio del trattamento con plasmaferesi non ha influenzato il tempo di risoluzione o la gravità dei sintomi all’inizio. Tuttavia, un caso clinico indica che la plasmaferesi è indicata quando è presente una profonda atassia, compromissione motoria e respiratoria.I pazienti devono essere monitorati attentamente per il rischio di sviluppare gravi complicanze sistemiche come insufficienza respiratoria superiore. Gli studi non mostrano alcun miglioramento significativo nel tempo di recupero con il trattamento IV IgG o plasmaferesi in MFS, quindi il monitoraggio può essere l’opzione migliore di un professionista.
Mentre raro, la condizione non deve andare non diagnosticata. Riconoscendo le caratteristiche cliniche chiave, i professionisti possono rivelare la causa dei sintomi del loro paziente e acquisire fiducia in un recupero completo.
Drs. Wang e Cantrell sono optometristi del personale presso l’Orlando VA Medical Center. Dr. Cali è un optometrista personale presso la Lee County VA Clinic. Gli autori desiderano ringraziare Joseph Miller, OD, Paul Gruosso, OD, e Vanessa Santos, OD, per l’assistenza con questo manoscritto.
1. Collier J. Neurite periferica: Le lezioni di Morisa. Edimburgo Med J. 1932:39: 601-18.
2. Fisher M. Una variante insolita di polineurite idiopatica acuta (sindrome oftalmoplegia, atassia e areflessia). N Ing J Med. 1956:255:57-65.
3. LoL, et al. Spettro clinico e immunologico della sindrome di Miller Fisher. Nervo muscolare. 2007:36(5):615-27.
4. Mori M, Kuwabara S, Fukutake T, et al. Caratteristiche cliniche e prognosi della sindrome di Miller Fisher. Neurologia. 2001:56(8): 1105-6.
5. Mori M, Kuwabara S, Fukutake T, et al. Plasmafaresi e sindrome di Miller Fisher: Analisi di 50 casi consecutivi. J Neurol Neurochirurgia Psych. 2002:72:680.
6. Il suo nome è Yuan Sindrome di Miller Fisher: uno studio retrospettivo basato sull’ospedale. Euro J Neurol. 2000:44(2):79-85.
7. Per maggiori informazioni:
8. Berlit P, Rakicky J. La sindrome di Miller Fisher. Revisione della letteratura. J neuro-oftalmologia clinica. 1992:12:57-63.
9. Il suo nome deriva da Ly Sindrome di Guillain-Barré a Taiwan: uno studio clinico su 167 pazienti. J Neurol Neurochirurgia Psych. 1997:63:494-500.
10. Tatsomoto M, Odaka M, Hirata K, Yuki N. Isolato abducens paralisi come variante regionale della sindrome di Guillain-Barré. J Neurol Sci. 2006:243:35-8.
11. Weiss JA, JC Bianco. Correlazione della conduzione afferente 1a con atassia della sindrome di Fisher. Nervo muscolare. 1986:9:327-32.
12. D. P., Biswash S, et al. Variante di Miller Fisher della sindrome di guillain-barré: un caso clinico e una revisione clinica. Bangabandhu Sheikh Mujib Medical Univer J. 2012: 5 (1): 69-71
13. Kornberg AJ, Pestronk A, Blume GM, et al. Colorazione selettiva dello strato molecolare cerebellare mediante IgG sieriche di Miller Fisher e sindromi correlate. Neurologia. 1996:47:1317-20.
14. Inoue N, Koh C, Iwahashi T. Rilevazione di anticorpi anticerebellari sierici in pazienti con sindrome di Miller Fisher. Euro J Neurol. 1999:42:230-4.
15. Barontini F, Di Lollo S, Maurri S, Lambruschini P. Localizzazione del processo patologico nella sindrome di Miller Fisher. Italia J Neurol Sci. 1992:13:221-5.
16. Il suo nome deriva dal latino “Koga M”, “Yuki N”, “Takahashi M”, et al. Stretta associazione di anticorpi anti-gangliosidici IgA con infezione antecedente Campylobacter jejuni nelle sindromi di Guillain-Barré e Fisher. J Neuroimmunolo. 2003:141:112-7.
17. Guiloff RJ. Conduzione nervosa periferica nella sindrome di Miller Fisher. J Neurol Neurochirurgia Psychiat. 1977:40:801-7.
18. Chikakiya H, Kunishige M, Yoshino H, et al. Neuropatia motoria e sensoriale ritardata in un paziente con encefalite del tronco cerebrale. J Neurol Sci. 2005:234:105-8.
19. Arakata Y, Yoshimuro M, Kobayashi S, et al. L’uso di immunoglobulina endovenosa nella sindrome di Miller Fisher. Il cervello si sviluppa J. 1993:15:231-3.
20. Il suo nome deriva dal greco antico, che significa “terra”, “terra”, “terra”, “terra”, “terra” e “terra”. Sindrome di Miller Fisher con coma transitorio: confronto con encefalite del tronco cerebrale di bickerstaff. Cervello Sviluppare J. 2002: 24: 98-101.
21. Yeh JH, Chen WH, Chen JR, et al. Sindrome di Miller Fisher con coinvolgimento centrale: trattamento di successo con plasmaferesi. Therap Aferesi dialisi J. 1999:3: 69-71.
22. Papanikolaou T, Cath G, Boothman B, et al. Case report: oftalmoparesi bilaterale acuta con midriasi areflessica pupillare nella sindrome di Miller-Fisher trattata con immunoglobulina endovenosa. Ophthalmol. 2010; ID articolo 291840.
23. Sandler RD, Hoggard N, Hadjivassiliou M. Sindrome di Miller-Fisher: L’atassia è centrale o periferica? Atassie del cervelletto. 2015:2:3.
24. Ejma M, Waliszewska-Prosół M, Hofman A, et al. Sindrome di Miller-fisher subacuta progressiva trattata con successo con plasmaferesi. Polacco J Neurol Neurochirurgia. 2015;49(2):137-8.
25. San-Juan OD, Martínez-Herrera JF, Moreno García J, et al. Sindrome di Miller Fisher: 10 anni di esperienza in un centro di terzo livello. Euro Neurol. 2009; 62:149-54
26. Snyder LA, Rismondo V, Miller NR. La variante di Fisher della sindrome di Guillain-Barré (sindrome di Fisher). J Neurooftalmolo. 2009 Dicembre; 29 (4): 312-24.
27. It M, Kuwabara S, Odaka M, et al. L’encefalite del tronco cerebrale di Bickerstaff e la sindrome di fisher formano uno spettro continuo: analisi clinica di 581 casi. J Neurol. 2008;255:674-82.
28. Yuki N. Paresi acuta dei muscoli extraoculari associati all’anticorpo IgG anti-GQ1b. Ann Neurol 1996;39: 668-72.