Faible aux genoux: Syndrome de Miller Fisher
Le syndrome de Miller Fisher (MFS), une variante rare et auto-limitante du syndrome de Guillain-Barré (SGB), est un syndrome d’anticorps anti-GQ1bIgG qui affecte les systèmes nerveux périphérique et central.1 Condition polyneuropathique neuromusculaire aiguë, la MFS provoque une paralysie ascendante avec une triade de présentation classique d’ophtalmoplégie, d’ataxie et d’aréflexie. Les chercheurs ont décrit la triade clinique pour la première fois en 1932.1 Deux décennies plus tard, le spécialiste canadien de l’AVC Charles Miller Fisher a été le premier à publier un rapport en 1956 qui décrivait les résultats cliniques et définissait la maladie comme une forme limitée de SGB.2
Les symptômes surviennent sur plusieurs jours, souvent en hiver et au printemps, généralement à la suite d’une infection virale, et les patients présentent souvent une diplopie comme l’un des premiers symptômes. Lorsqu’un patient présente des symptômes tels qu’une ésotropie intermittente, une poursuite horizontale altérée et des pupilles mydriatiques fixes, et que votre examen neurologique est anormal, il est temps de penser au SGB, en particulier à la MFS. Le cas suivant illustre les signes et symptômes pertinents et reflète le résultat typique.
Antécédents
Un homme de race blanche de 27 ans s’est présenté à la Clinique ophtalmologique de l’AV au cours des mois de printemps, signalant une diplopie binoculaire aiguë qui avait commencé la veille. Le patient a déclaré qu’il était constant, mais qu’il ne se produisait qu’à distance avec des objets déplacés horizontalement. Il n’y avait pas de douleur oculaire ou de vision trouble associée à la diplopie. Il s’est également plaint de vertiges récents et de difficultés d’équilibre. Il a signalé une infection des sinus avec une congestion paranasale importante qui a commencé une semaine avant la double vision. Les antécédents oculaires et médicaux antérieurs n’étaient pas remarquables; le patient ne prenait actuellement aucun médicament et n’avait aucune allergie médicamenteuse.
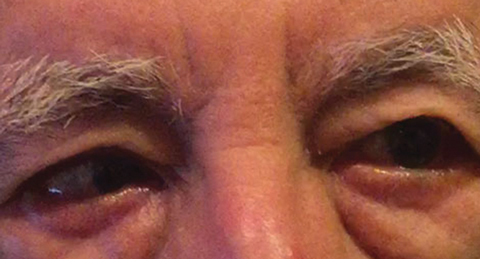
Fig. 1. Le diagnostic différentiel après l’examen initial comprenait une paralysie partielle du CN VI droit, ce qui pourrait indiquer une condition vasculaire, comme on l’a vu ici chez un autre patient. Cliquez sur l’image pour l’agrandir. Photo: Michael DelGiodice, DO
Données diagnostiques
Lors de l’examen de base, les acuités visuelles non corrigées étaient de 20/20-2 DO et de 20/25-2 OS. Les pupilles étaient de 5 mm également rondes et réactives à la lumière, sans défaut pupillaire afférent. Les mouvements extraoculaires étaient pleins, lisses et précis dans les deux yeux sans aucune restriction. Le test de couverture à distance a révélé une ésotropie droite dioptrique intermittente de quatre à six prismes dans le regard primaire se produisant environ 50% du temps. L’exécution d’un test de couverture à near a révélé une œsophorie dioptrique à quatre prismes dans le regard primaire sans tropie.
Bien qu’ils ne soient pas entrepris dans ce cas, les cliniciens doivent couvrir le test en regard primaire, droit et gauche. Chez les patients atteints de diplopie, la paralysie du nerf crânien six (CN VI) se manifestera par une déviation non comitante, pire dans le regard droit ou gauche, selon le côté de la paralysie. Une phorie décompensante montrera, en revanche, une déviation comitante.
L’erreur de réfraction était de -0,50 D sphère OD et de -0,75D sphère OS et la vision était corrigible à 20/20 dans chaque œil. Les pressions intraoculaires étaient dans les limites normales des deux yeux. Les résultats des segments antérieur et postérieur n’étaient pas remarquables dans les yeux droit et gauche. Les nerfs optiques semblaient en bonne santé avec un rapport tasse/ disque de 0,15 dans les deux yeux.
Les diagnostics différentiels comprenaient une paralysie partielle du CN VI droit (Figure 1), une myasthénie grave, un processus de compression et une ésophorie de distance décompensante provoquant une ésotropie droite intermittente. Le patient a nié tout antécédent de diplopie binoculaire.
L’imagerie du cerveau et des orbites a été ordonnée par la clinique ophtalmologique pour exclure toute atteinte cérébrale, y compris une lésion compressive. La tomodensitométrie (TDM) du cerveau et des orbites a été réalisée et interprétée par le neuroradiologue du personnel. La tomodensitométrie a été choisie au lieu de l’imagerie par résonance magnétique (IRM) en raison du moment et de la disponibilité; les tests et les résultats pouvaient être obtenus le même jour avec la tomodensitométrie à la clinique après les heures de travail. Les résultats n’étaient pas remarquables, aucun signe d’hémorragie intracrânienne, de lésion compressive ou d’infarctus aigu.
La tomodensitométrie des sinus paranasaux a révélé une maladie chronique des sinus paranasaux modérée à sévère et une opacification complète du sinus maxillaire droit. Il n’y avait pas d’extension dans l’orbite. Le médecin de famille du patient a été informé des symptômes du patient, des résultats de l’examen de la vue et des résultats de l’imagerie par tomodensitométrie et a commencé le traitement de la sinusite maxillaire avec une dose orale d’azithromycine de cinq jours et a demandé au patient de faire un suivi auprès de la clinique ophtalmologique.
Le patient est revenu cinq jours plus tard après avoir terminé son traitement par voie orale à l’azithromycine et a déclaré qu’il n’avait constaté aucune amélioration de la diplopie à distance; en outre, il a connu l’apparition d’une photophobie significative et d’une douleur périoculaire autour des deux yeux. Le patient a déclaré que son déséquilibre de la démarche et ses difficultés à marcher s’aggravaient également. À l’examen, les acuités visuelles les mieux corrigées sont restées de 20/20 dans les yeux droit et gauche. Les pupilles ont été dilatées à 8,5 mm sans aucune réactivité à la lumière dans les deux yeux (Figure 2). Les mouvements extraoculaires ont révélé une restriction latérale avec douleur lors du mouvement dans l’œil droit et une restriction médiale avec douleur lors du mouvement dans l’œil gauche compatible avec une paralysie du regard droit (Figure 3). Les activités horizontales et verticales étaient saccadées avec des mouvements imprécis et saccadés. Les résultats des segments antérieur et postérieur sont restés peu remarquables.
Les diagnostics différentiels après examen ophtalmologique comprenaient une ophtalmoplégie internucléaire gauche, une paralysie du regard droit et une paralysie du regard droit avec une ophtalmoplégie internucléaire gauche, ce qui pourrait expliquer les résultats de la motilité extraoculaire. D’autres différences incluent une lésion du mésencéphale ou une myasthénie grave. Les diagnostics différentiels de dilatation de la pupille comprennent une paralysie du troisième nerf (CN III), un traumatisme et une dilatation pharmacologique. Le patient n’a présenté aucun autre résultat compatible avec une troisième paralysie nerveuse et a nié un traumatisme ou une exposition à des agents pharmacologiques.
Le patient a été dirigé vers la neurologie pour une évaluation plus approfondie. L’examen neurologique a révélé que le patient était complètement alerte et orienté sans perte de sensation faciale. Le patient présentait une ataxie légère avec des difficultés à marcher en tandem et à se tenir debout en position accroupie. Une faiblesse proximale des membres supérieurs et inférieurs et des réflexes anormaux étaient présents. Toutes les modalités sensorielles étaient intactes. L’IRM et l’angiographie par résonance magnétique (ARM) du cerveau et des orbites n’étaient pas remarquables. La protéine du liquide céphalo-rachidien a été élevée à 75 mg / dL (plage de référence: 15 mg / DL à 45 mg / DL) sans autre anomalie. Les tests d’électromyogramme n’étaient pas remarquables et n’ont montré aucune anomalie de la conduction nerveuse de la colonne vertébrale aux pieds et aux mains.
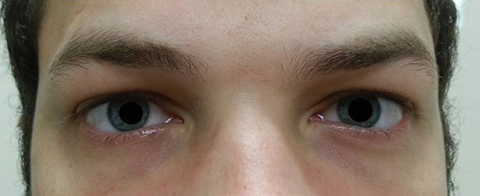
Fig. 2. Les pupilles du patient ont été dilatées de 8,5 mm sans réactivité dans les yeux droit ou gauche. Cliquez sur l’image pour l’agrandir.
Diagnostic
Le patient présentait des symptômes aigus de diplopie, d’ataxie et d’aréflexie. Compte tenu de l’ésotropie droite intermittente, des poursuites horizontales altérées, de la pupille mydriatique fixe, d’une ataxie légère et de réflexes anormaux à l’examen neurologique et d’une élévation de la protéine du liquide céphalo-rachidien, le diagnostic de MFS a été posé. Le patient a été admis à l’hôpital pour surveiller toute autre complication. Un patch oculaire a été fourni au patient pour couvrir l’œil droit au besoin pour la diplopie.
Aucun critère diagnostique publié pour le syndrome de Miller Fisher n’existe actuellement. Le diagnostic est généralement posé par la présentation de la triade clinique ainsi que par des études d’imagerie et de liquide céphalo-rachidien. L’IRM est généralement banale dans la condition, bien que certaines études aient montré des lésions centrales dans le mésencéphale, le pons et la médullaire inférieure. Les études électrophysiologiques montrent une conduction sensorielle périphérique réduite ou anormale.17
La protéine du liquide céphalo-rachidien est souvent élevée sans autre résultat anormal. Dans une étude historique, 64,4% des patients atteints de MFS présentaient une teneur élevée en protéines du liquide céphalo-rachidien.8 Un test d’anticorps IgG anti-GQ1b positif permet un diagnostic définitif. Dans une étude, l’anticorps anti-IgG GQ1b était présent chez jusqu’à 95% des patients atteints de la maladie et absent chez les témoins.3
Le neurologue a diagnostiqué au patient une MFS basée sur la triade clinique d’ophtalmoplégie, d’ataxie et d’aréflexie, une IRM et une ARM du cerveau banales et une protéine élevée du liquide céphalo-rachidien. Par conséquent, aucun test d’anticorps anti-IgG GQ1b n’a été effectué dans ce cas.
Plusieurs caractéristiques cliniques de la MFS sont également observées dans le syndrome de Guillain-Barré et l’encéphalite du tronc cérébral de Bickerstaff, ce qui rend le diagnostic difficile. Les patients atteints uniquement de Guillain-Barré présentent une faiblesse des membres, une perte sensorielle, une neuropathie crânienne et une aréflexie sans manifestations ophtalmologiques. Les patients atteints d’encéphalite du tronc cérébral de Bickerstaff présentent généralement une ophtalmoplégie, une ataxie, une hyper-réflexie et une conscience perturbée. Étant donné que plusieurs symptômes et signes sont présents dans les trois conditions, le test du titre d’anticorps IgG anti-GQ1b est utile pour un diagnostic plus définitif. L’anticorps anti-IgG GQ1b est positif chez une grande majorité des patients atteints du syndrome de Miller Fisher, contre 66% chez l’encéphalite du tronc cérébral de Bickerstaff et 26% chez les patients atteints du syndrome de Guillain-Barré.16
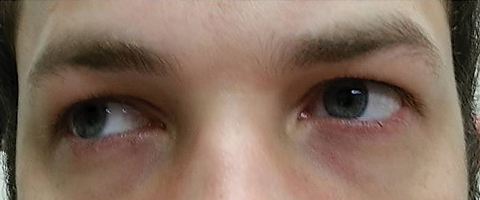
Fig. 3. Les mouvements extraoculaires ont révélé une restriction de la dextroversion. Cliquez sur l’image pour l’agrandir.
Traitement et suivi
Le patient a été évalué le lendemain de son admission, lorsqu’il a déclaré n’avoir subi aucun changement visuel ou amélioration de l’équilibre. La diplopie binoculaire était toujours constante; par conséquent, l’œil droit restait rapiécé. Des douleurs oculaires avec des regards lointains et des mouvements oculaires rapides étaient toujours présentes. Lors d’un examen limité au chevet du patient, les acuités visuelles les mieux corrigées sont restées de 20/20 dans les yeux droit et gauche. Les pupilles ont été dilatées à 8 ans.0 mm dans les deux yeux avec une réactivité minimale, légèrement améliorée par rapport à la veille. La vision des couleurs était normale dans les yeux droit et gauche. Les mouvements extraoculaires étaient pleins dans tous les quadrants avec une douleur encore présente sur les regards horizontaux lointains. Les activités horizontales sont restées saccadées avec des mouvements saccadés et un nystagmus terminal. Les nerfs optiques semblaient sains avec des marges distinctes et un rapport tasse / disque de 0,15 dans les deux yeux.
Deux jours après, le patient a déclaré une amélioration de tous les symptômes et que ses yeux « se sentent mieux aujourd’hui qu’ils ne l’ont été depuis un certain temps. »La douleur oculaire et la diplopie binoculaire ont toutes deux été résolues. Les acuités visuelles sont restées 20/20 dans les yeux droit et gauche. La taille des pupilles était de 7,5 mm dans l’obscurité et de 7,0 mm dans la lumière, avec des réponses directes et consensuelles améliorées dans les deux yeux. Les mouvements extraoculaires étaient pleins dans tous les quadrants sans douleur oculaire. Les activités horizontales sont restées saccadées avec des mouvements saccadés et un nystagmus terminal. L’examen neurologique a révélé des réflexes anormaux, qui sont restés stables. Les options de traitement de la MFS comprennent la plasmaphérèse et les IgG d’immunoglobulines intraveineuses (IV), mais celles-ci ont été retenues compte tenu de l’amélioration de l’ophtalmoplégie et de l’ataxie.
Quatre jours plus tard, le patient a signalé que tous les symptômes s’amélioraient encore, avec quelques difficultés à se concentrer et à suivre lors de l’utilisation de l’ordinateur. L’équilibre était résolu à 80% et il y avait peu d’occurrences de diplopie à distance. Les acuités sont restées 20/20 OU. Les pupilles étaient de 5,5 mm dans l’obscurité et de 4,0 mm dans la lumière avec 3+ réponses directes et consensuelles OU. Les mouvements extraoculaires étaient pleins dans tous les quadrants sans douleur oculaire. Les activités horizontales ont été améliorées, sans mouvements saccadiques. L’accommodation monoculaire était normale, ce qui a été mesuré à 8,5 et 9 dioptries de prisme OD et OS, respectivement.
Une semaine plus tard, le patient a signalé que sa vision était revenue à la normale, sans diplopie et avec de légères difficultés de mise au point et de suivi. L’équilibre a été résolu à 100%. Les acuités visuelles sont restées 20/20 dans les yeux droit et gauche. La taille des pupilles était de 5,5 mm dans l’obscurité, 4,0 mm dans la lumière avec 4+ réponses directes et consensuelles OU. Les mouvements extraoculaires étaient pleins dans tous les quadrants sans douleur oculaire. Les activités horizontales se sont améliorées sans mouvements saccadiques observés. La santé oculaire était banale.
Le traitement a été interrompu compte tenu de la résolution de l’ophtalmoplégie et de l’ataxie avec aréflexie stable. Le rétablissement complet de l’ophtalmoplégie et de l’ataxie a pris deux semaines. La prise en charge comprenait une surveillance étroite et un suivi en quatre semaines; on a conseillé au patient de revenir plus tôt si l’un de ses symptômes revenait.
Caractéristiques cliniques
Dans des études bien décrites sur la MFS, tous les patients présentaient la triade clinique d’ophtalmoplégie, d’ataxie et d’aréflexie.4,5 L’apparition des symptômes se produit généralement sur plusieurs jours et les patients souffrent d’une infection virale une à quatre semaines avant l’apparition des symptômes cliniques.4,5 Selon une étude de 50 patients atteints de la maladie, l’intervalle moyen entre le début de l’infection et le développement des symptômes neurologiques est de huit jours.4 Les autres signes de MFS comprennent des troubles de l’élocution, des difficultés à avaler et une expression faciale anormale avec une incapacité à sourire ou à siffler.
Caractéristiques ophtalmologiques. Les praticiens doivent être à l’affût de la mydriase, de la rétraction du couvercle, de la fermeture de l’angle aigu et de la diplopie secondaire à une paralysie nerveuse affectant les CN III, IV et VI.3,8,10,22,24 La diplopie est le symptôme ophtalmologique le plus courant rapporté dans de nombreuses études sur la MFS.8,16,25,26,27 La diplopie est également le symptôme initial rapporté chez 38,6% des 223 patients dans une étude historique et 65% des 466 patients dans une deuxième étude.8,27 Dans la deuxième étude, 100% des patients présentaient une ophtalmoplégie externe, tandis qu’une ophtalmoplégie interne était présente chez 35% des patients.27 L’ophtalmoplégie externe fait référence à une altération des muscles extraoculaires externes. L’ophtalmoplégie interne se réfère à une altération du sphincter pupillaire et du muscle ciliaire. L’ophtalmoplégie complète affecte à la fois les muscles externes et internes.
Dans une étude rétrospective de 19 patients atteints de MFS, tous présentaient une ou plusieurs paralysie nerveuse affectant les muscles extraoculaires.25 Autres études montrent que les patients atteints de MFS peuvent présenter plusieurs paralysies du nerf crânien, qui peuvent se manifester de manière bilatérale.22,24,25 Un résultat commun trouvé parmi les troubles positifs aux IgG anti-GQ1b est un déficit d’abduction compatible avec une paralysie CN VI unilatérale ou bilatérale.26,28 L’ophtalmoplégie interne provoque une mydriase de la pupille, où la constriction de la pupille à la lumière et / ou une stimulation proche peut aller de minimale à absente.8,26 Dans un rapport, la mydriase était présente chez 42% des patients. L’atteinte de CN VII est survenue chez environ 45,7% des patients atteints de MFS, provoquant une faiblesse faciale, une incapacité à sourire ou à siffler.8
Ataxie. La cause de cette caractéristique clinique dans la MFS n’est pas entièrement comprise. Le débat existe pour savoir si l’ataxie est causée par un dysfonctionnement central du cervelet ou périphérique. La première hypothèse, proposée par le Dr Fisher, postule l’implication de neurones Ia-afférents.2 Ceci a été corroboré par une deuxième étude des décennies plus tard, qui a montré des anomalies des fibres Ia-afférentes dans le MFS.11 Une atteinte nerveuse périphérique anormale a également été proposée avec une inadéquation entre l’entrée proprioceptive des broches musculaires et l’information kinesthésique des récepteurs articulaires.12
Il existe également des preuves à l’appui que l’ataxie provient du cervelet. Une prédominance de l’anticorps anti-IgG GQ1b dans le cervelet a été trouvée à l’aide d’une coloration immunocytochimique du cervelet humain et confirmée par des études de western blot, qui ont montré une augmentation de l’anticorps anti-IgG GQ1b chez les patients atteints de MFS par rapport au contrôle, bien que le mécanisme d’atteinte cérébelleuse n’ait pas été étudié.13,14 En 2015, les chercheurs ont publié un rapport de cas d’un patient MFS qui a subi une IRM. Il y avait une réduction du rapport N-acétylaspartate / créatine, suggérant un dysfonctionnement cérébelleux, qui est revenu à la normale 2,5 mois après la récupération.23
Aréflexie. Cette caractéristique clinique, associée à une dépression des réflexes tendineux profonds, est un signe d’implication du système nerveux périphérique dans la MFS. Des études électrophysiologiques ont validé une conduction nerveuse périphérique anormale dans le SGB et le MFS.3 Dans une étude, 81,6% des cas de SSM présentaient une fexie.8 Une autre étude historique a montré que tous les patients présentaient une aréflexie, qui était toujours présente six mois après son apparition.4
À quel point le MFS est-il courant?
Les données épidémiologiques sur le syndrome de Miller Fisher sont limitées. L’incidence de la MFS est assez rare, se produisant dans 0,09 pour 100 000 personnes par an.3 MFS survient plus fréquemment chez les patients atteints du syndrome de Guillain-Barré, de 3% à 25%.4,6,7 Cette association se produit davantage chez les patients d’origine extrême-orientale, suggérant une composante génétique possible de la MFS. L’âge moyen d’apparition est de 43 ans, avec une plage de 13 à 78,5 MFS affecte les mâles deux fois plus que les femelles avec un rapport de deux à 1,03,5 MFS se produit davantage en hiver et au printemps.5,6,8 La cause exacte de la prédilection saisonnière n’a pas été déterminée, mais elle est probablement associée à la nature post-infectieuse de la maladie, car des infections bactériennes et virales déclenchent une réponse auto-immune provoquant la production de l’anticorps anti-GQ1b IgG dans la MFS.
Physiopathologie
La physiopathologie du syndrome de Miller Fisher n’est pas bien comprise; cependant, plusieurs hypothèses existent à partir d’études immunologiques et histologiques. Il est bien connu que la maladie se situe dans le spectre des syndromes d’anticorps anti-IgG GQ1b avec le syndrome de Guillain-Barré et l’encéphalite du tronc cérébral de Bickerstaff.
Le ganglioside GQ1b est un groupe de lipides complexes impliqués dans les systèmes nerveux central et périphérique. GQ1b est un composant de la structure de la membrane plasmique des nerfs crâniens qui alimentent les muscles extraoculaires et est impliqué dans la fonction cellulaire au niveau de la jonction neuromusculaire présynaptique.
Un mécanisme auto-immun issu d’une infection déclenchante précédente produira l’anticorps IgG anti-GQ1b, qui endommage la fonction ganglioside GQ1b, provoquant une démyélinisation. Des études histologiques ont montré une démyélinisation et un gonflement axonal des nerfs crâniens périphériques et oculomoteurs.15 L’anticorps anti-GQ1bIgG est absent chez les patients en bonne santé, mais positif chez plus de 90% des patients atteints de MFS.16,25
Des infections bactériennes et virales déclenchent une réponse auto-immune, provoquant la production de l’anticorps anti-GQ 1b IgG. Les agents infectieux suivants qui ont été associés à la MFS comprennent la pneumonie à mycoplasmes, le VIH, Campylobacter jejuni, Hemophilus influenza, Helicobacter pylori et le virus d’Epstein-Barr.4,16 Dans un rapport portant sur 466 patients atteints de SSM, 90% avaient une maladie antérieure, y compris une infection des voies respiratoires supérieures, une diarrhée ou les deux.27
Pronostic et traitement
Le syndrome de Miller Fisher est une maladie auto-limitante et a un pronostic globalement positif. Les symptômes s’améliorent généralement après quelques semaines, la guérison complète se produisant généralement en deux à trois mois. Dans une étude, la récupération a commencé à une médiane de 13 jours après l’apparition des symptômes, et la résolution complète de l’ophtalmoplégie et de l’ataxie s’est produite en six mois.4,5 Rechutes ont été constatées dans moins de 3% des cas.4,5
Bien que la SSM soit généralement auto-limitante, des complications systémiques ont été associées à la maladie. Des infections des voies respiratoires supérieures ont été trouvées chez 56% à 76% des patients atteints de MFS, pouvant évoluer vers une insuffisance respiratoire nécessitant une ventilation mécanique.4,18,19 D’autres complications graves rares comprennent la cardiomyopathie, l’acidose lactique et le coma.3,20
Les options de traitement de la MFS comprennent la plasmaphérèse, les IgG d’immunoglobulines IV et la surveillance sans traitement. Les chercheurs postulent que la plasmaphérèse et les IgG IV peuvent être efficaces pour accélérer le temps de résolution puisque l’anticorps Gq1b est une IgG de classe et que la demi-vie des IgG est d’environ 21 jours, ce qui est plus long que les demi-vies de cinq à six jours des IgM et des IgA.19 Cependant, aucune étude randomisée à double insu contrôlée contre placebo portant sur ces traitements n’a été menée.
Une étude rétrospective n’a pas montré que la plasmaphérèse modifiait les chances de guérison complète et le temps de résolution de l’ataxie et de l’ophtalmoplégie; de plus, l’intervalle entre le début et le début du traitement par plasmaphérèse n’affectait pas le temps de résolution ni la gravité des symptômes au début. Cependant, un rapport de cas indique que la plasmaphérèse est indiquée en cas d’ataxie profonde, d’insuffisance motrice et respiratoire.21
Les patients doivent être surveillés de près pour le risque de développer des complications systémiques graves telles qu’une insuffisance respiratoire supérieure. Les études ne montrent aucune amélioration significative du temps de récupération avec les IgG IV ou le traitement par plasmaphérèse dans la MFS, la surveillance peut donc être la meilleure option pour un praticien.
Bien que rare, la maladie n’a pas besoin d’être diagnostiquée. En reconnaissant les principales caractéristiques cliniques, les praticiens peuvent révéler la cause des symptômes de leur patient et gagner en confiance dans un rétablissement complet.
Les Drs Wang et Cantrell sont optométristes au Orlando VA Medical Center. Le Dr Cali est optométriste à la clinique Lee County VA. Les auteurs tiennent à remercier Joseph Miller, DO, Paul Gruosso, DO, et Vanessa Santos, DO, pour leur aide dans la rédaction de ce manuscrit.
1. Collier J. Névrite périphérique: Les conférences Morisa. Edinburgh Med J. 1932:39:601-18.
2. Fisher M. Une variante inhabituelle de la polynévrite idiopathique aiguë (syndrome ophtalmoplégie, ataxie et aréflexie). N Eng J Med. 1956:255:57-65.
3. Lo YL, et coll. Spectre clinique et immunologique du syndrome de Miller Fisher. Nerf musculaire. 2007:36(5):615-27.
4. Mori M, Kuwabara S, Fukutake T, et al. Caractéristiques cliniques et pronostic du syndrome de Miller Fisher. Neurologie. 2001:56(8): 1105-6.
5. Mori M, Kuwabara S, Fukutake T, et al. Plasmapharésie et syndrome de Miller Fisher: Analyse de 50 cas consécutifs. J Neurol Neurosurg Psych. 2002:72:680.
6. Yuans CL, Wang YJ, Tsai CP. Syndrome de Miller Fisher: Une étude rétrospective en milieu hospitalier. Euro J Neurol. 2000:44(2):79-85.
7. Cheng BC, Chang WN, Chang CS et al. Syndrome de Guillain-Barré dans le sud de Taiwan: Caractéristiques cliniques, facteurs pronostiques et résultats thérapeutiques. Euro J Neurol. 2003:10 (6):655-62.
8. Berlit P, Rakicky J. Le syndrome de Miller Fisher. Revue de la littérature. J neuro-ophtalmologie clinique. 1992:12:57-63.
9. Lyu RK, Tang LM, Cheng SY, et al. Syndrome de Guillain-Barré à Taiwan: Une étude clinique de 167 patients. J Neurol Neurosurg Psych. 1997:63:494-500.
10. Tatsomoto M, Odaka M, Hirata K, Yuki N. A isolé la paralysie d’abducens comme variante régionale du syndrome de Guillain-Barré. J Neurol Sci. 2006:243:35-8.
11. Weiss JA, JC blanc. Corrélation de la conduction afférente 1a avec l’ataxie du syndrome de Fisher. Nerf musculaire. 1986:9:327-32.
12. Das P, Biswash S, et coll. Variante de Miller Fisher du syndrome de guillain-barré: Rapport de cas et examen clinique. Université médicale de Bangabandhu Sheikh Mujib J. 2012:5 (1): 69-71
13. Kornberg AJ, Pestronk A, Blume GM, et al. Coloration sélective de la couche moléculaire cérébelleuse par les IgG sériques de Miller Fisher et les syndromes apparentés. Neurologie. 1996:47:1317-20.
14. Inoue N, Koh C, Iwahashi T. Détection d’anticorps anticérébelleux sériques chez des patients atteints du syndrome de Miller Fisher. Euro J Neurol. 1999:42:230-4.
15. Barontini F, Di Lollo S, Maurri S, Lambruschini P. Localisation du processus pathologique dans le syndrome de Miller Fisher. Italie J Neurol Sci. 1992:13:221-5.
16. Koga M, Yuki N, Takahashi M, et al. Association étroite des anticorps anti-gangliosides IgA avec une infection antérieure à Campylobacter jejuni dans les syndromes de Guillain-Barré et de Fisher. J Neuroimmunol. 2003:141:112-7.
17. Guiloff RJ. Conduction nerveuse périphérique dans le syndrome de Miller Fisher. J Neurol Neurochirurgie Psychique. 1977:40:801-7.
18. Chikakiya H, Kunishige M, Yoshino H, et al. Neuropathie motrice et sensorielle retardée chez un patient atteint d’encéphalite du tronc cérébral. J Neurol Sci. 2005:234:105-8.
19. Arakata Y, Yoshimuro M, Kobayashi S, et al. L’utilisation d’immunoglobulines intraveineuses dans le syndrome de Miller Fisher. Développement du cerveau J. 1993:15:231-3.
20. Matsumoto H, Kobayashi O, Tamura K, et al. Syndrome de Miller Fisher avec coma transitoire: comparaison avec l’encéphalite du tronc cérébral de bickerstaff. Développement du cerveau J. 2002:24:98-101.
21. Yeh JH, Chen WH, Chen JR, et al. Syndrome de Miller Fisher avec implication centrale: traitement réussi par plasmaphérèse. Dialyse par aphérèse Therap J.1999:3:69-71.
22. Papanikolaou T, Cath G, Boothman B, et al. Rapport de cas: ophtalmoparésie bilatérale aiguë avec mydriase aréflexique pupillaire dans le syndrome de Miller-Fisher traitée par immunoglobuline intraveineuse. J Ophthalmol. 2010; Numéro D’article 291840.
23. Sandler RD, Hoggard N, Hadjivassiliou Syndrome de M. Miller-Fisher: L’ataxie est-elle centrale ou périphérique? Ataxies du cervelet. 2015:2:3.
24. Ejma M, Waliszewska-Prosół M, Hofman A, et al. Syndrome subaiguë progressif de Miller-fisher traité avec succès par plasmaphérèse. Neurochirurgie polonaise J Neurol. 2015;49(2):137-8.
25. San-Juan OD, Martínez-Herrera JF, Moreno García J, et al. Syndrome de Miller Fisher: 10 ans d’expérience dans un centre de troisième niveau. Euro Neurol. 2009; 62:149-54
26. Snyder LA, Rismondo V, Miller NR. La variante de Fisher du syndrome de Guillain-Barré (syndrome de Fisher). J Neuroophtalmol. 2009 Déc; 29(4): 312-24.
27. Ito M, Kuwabara S, Odaka M, et al. L’encéphalite du tronc cérébral de Bickerstaff et le syndrome de Fisher forment un spectre continu: analyse clinique de 581 cas. J Neurol. 2008;255:674-82.
28. Yuki N. Parésie aiguë des muscles extraoculaires associée à l’anticorps IgG anti-GQ1b. Ann Neurol 1996; 39:668-72.