Conception de sondes FRET-TaqMan pour PCR en temps réel multiplex utilisant un contrôle positif interne
Introduction
La PCR en temps réel permet une surveillance continue de la fluorescence des fluorophores lors de la génération de produits de PCR dans un format de tube fermé. Actuellement, les méthodes disponibles utilisent des sondes marquées ou un colorant d’intercalation de l’ADN pour surveiller l’amplification du produit de PCR. La sonde TaqMan est un oligonucléotide monocaténaire contenant un fluorophore et un extincteur séparés de 10 à 30 bases. La sonde TaqMan est hydrolysée pendant l’amplification en raison de l’activité exonucléase spécifique à double brin 5′-3′ de la polymérase Taq qui sépare le fluorophore de l’extincteur, ce qui entraîne une augmentation de la fluorescence. Les instruments de PCR en temps réel sont équipés de détecteurs de fluorescence et d’un logiciel capable d’estimer le seuil de cycle (Ct), le cycle auquel la fluorescence est supérieure à la fluorescence de fond, pour des réactions positives. Cependant, la technique ne peut pas différencier un vrai résultat négatif d’un faux négatif lorsque la PCR est affectée par des inhibiteurs d’amplification. Après l’extraction de l’acide nucléique, il est rapporté que des inhibiteurs peuvent encore être présents dans des échantillons cliniques (par exemple, l’hémoglobine), des échantillons environnementaux (par exemple, les acides humique et fulvique) et des produits chimiques utilisés lors de l’extraction de l’acide nucléique (par exemple, l’éthanol, les détergents ou les agents chaotropes). La fiabilité des tests diagnostiques est augmentée par l’inclusion d’un acide nucléique de contrôle interne qui peut indiquer la présence et l’impact d’inhibiteurs de PCR (1,2). Un contrôle positif interne (IPC) est amplifié simultanément en présence d’une séquence cible à l’aide d’un fluorophore marqué qui émet de la lumière à une longueur d’onde différente de celle du fluorophore utilisé pour le dosage de la séquence cible, les deux fluorophores étant détectés dans des canaux différents par l’instrument de PCR en temps réel. Le contrôle interne couramment utilisé pour la PCR est un plasmide qui contient une séquence similaire à celle de la cible d’essai, à l’exception de la région de la sonde. Un nombre limité de molécules IPC sont ajoutées à la cible d’essai individuelle et co-amplifiées avec l’acide nucléique cible; ainsi, un signal IPC positif est la preuve que la réaction d’amplification s’est suffisamment déroulée pour générer un signal positif à partir de très petites quantités d’acide nucléique cible. Cette caractéristique est importante pour assurer une amplification équivalente de la CIB et de l’acide nucléique cible (3-9). Cependant, certains dispositifs – tels que le LightCycler 1.2 et le LightCycler 2 (Roche Applied Science, Indianapolis, IN, USA), le Dispositif d’identification Avancé des agents Pathogènes Robuste (R.A.P.I.D.) instrument (Technologie Idaho, Salt Lake City, UT, États-Unis) et instruments PCR portables en temps réel — ne sont équipés que d’une source de lumière et d’un canal d’émission associé. Le multiplexage sur ces types d’instruments nécessite l’utilisation d’oligonucléotides multi-marqués et le concept de transfert d’énergie par résonance de fluorescence (FRET). Cependant, les sondes FRET peuvent être difficiles à concevoir et peuvent manquer de robustesse dans des régions hypervariables et lorsque des conditions d’amplification non optimales sont présentes. L’utilisation de sondes de FRET nécessite l’identification de deux régions pour deux sondes. Le système de sonde FRET repose sur une hybridation adjacente d’oligonucléotides des deux sondes mono-marquées de telle sorte que la fluorescence du colorant accepteur est détectée par l’excitation du fluorophore attaché à la deuxième sonde. Un inconvénient majeur de la sonde FRET est l’exigence d’une plus grande zone de séquence nécessaire pour accueillir deux sondes adjacentes avec un espace de 2 à 5 nucléotides. La possibilité existe pour le multiplexage avec d’autres systèmes d’étiquettes, tels que MegaStokes dye (DYXL; Dyomics, Jena, Allemagne) et Pulsar 650 (Biosearch Technologies, Inc., Novato, CA, USA), mais ces systèmes d’étiquetage nécessitent des informations spectrales pour compenser la diaphonie des canaux et ne sont pas largement disponibles. La sonde FRET-TaqMan développée dans cette étude ne nécessite cependant pas de logiciel de compensation de couleur et utilise des fluorophores (FAM, Cy5.5) largement disponibles. Le présent rapport se concentre sur la conception d’une sonde à triple marquage pour permettre le multiplexage dans des instruments équipés d’une seule source d’excitation à diode électroluminescente bleue (LED) (470 nm) et de trois canaux de détection correspondants associés (par exemple, 530 nm, 640 nm et 705 nm).
Matériaux et méthodes
Principe de conception de la sonde
Toutes les expériences ont été réalisées sur le système R.A.P.I.D. d’Idaho Technology, un instrument de PCR portable en temps réel à 32 échantillons équipé d’une LED bleue pour l’excitation (470 nm) et de trois canaux de détection de fluorescence associés qui mesurent la fluorescence à 530, 640 et 705 nm pour les dosages mono et bicolores. Les amorces JVAF et JVAR(10) et la sonde 5′-FAM/BHQ-1 ont été synthétisées à l’installation centrale de CDC Biotechnology (Atlanta, GA, USA) et la sonde 5’FAM-Cy5.5/3’BHQ-3 a été synthétisée à Idaho Technology. Pour permettre la PCR TaqMan duplex, une sonde a été marquée avec 5′-FAM/ BHQ-1 (longueur d’onde de fluorescence de 520 nm) et une autre avec 5’FAM-Cy5,5/3’BHQ-3 (longueur d’onde de fluorescence de 705 nm). En solution, la sonde marquée par 5’FAM-Cy5.5/3’BHQ-3 reste éteinte lors de l’hybridation à une séquence, et lors du clivage ultérieur de la sonde par extension d’amorce enzymatique, l’éteigneur est éliminé. Il en résulte un FRET par l’excitation de FAM par la source lumineuse de l’instrument R.A.P.I.D. L’énergie d’excitation est transférée au fluorophore accepteur, Cy5.5, et la fluorescence émise est mesurée dans un second canal de fluorescence (canal de fluorescence F3 pour l’instrument R.A.P.I.D.). L’émission de fluorescence Cy5.5 est le résultat de la combinaison des techniques de sonde FRET et TaqMan, de sorte que ces sondes à triple marquage peuvent être appelées sondes « FRET-TaqMan”.
Amorces, sondes et plasmide
Pour démontrer les capacités de multiplexage de la conception de FRET-TaqMan, nous avons utilisé un test de PCR en temps réel Cryptosporidium spécifique au genre pour amplifier une partie de 159 pb du gène de l’ARNr 18S (10). L’amorce avant (JVAF) était de 5′-ATGACGGGTAACGGGGAAT-3′, l’amorce inverse (JVAR) était de 5′-CCAATTACAAAAC-CAAAAAGTCC-3′ et la sonde TaqMan (JVAP) était de 5′-FAM-CGCGCCTGCT-GCCTTCCTTAGATG-BHQ1–3′, correspondant aux nucléotides 100-118, 258-236, et 184 à 161, respectivement, du numéro d’adhésion de GenBank. AY458612. Les amorces pour la construction du Cryptosporidium IPC contenaient le même ensemble de séquences d’amorces rapportées pour l’amplification de Cryptosporidium spp., à l’exception de la région de liaison à la sonde de l’ADN cible, qui a été remplacée par la séquence artificielle 5′-TTGTGCTCGTAGTCGCCTCTGTCTCT-3′ (JPIPC). Cette séquence de sondes avait une température de recuit de 69 ° C et était conçue pour ne pas avoir de structure secondaire. La spécificité théorique de la sonde interne a été déterminée par une recherche par SOUFFLE (11) des séquences de sonde et d’amorce dans laquelle aucune correspondance exacte n’a été trouvée avec une séquence de Cryptosporidium. Le fragment de CIB a été synthétisé et cloné en un plasmide par Integrated DNA Technologies (Coralville, IA, USA). Une concentration connue d’ADN plasmidique (100 copies/réaction) a été ajoutée dans le mélange maître de toutes les réactions sauf dans le témoin négatif. Une sonde à triple marquage (JPIPC) spécifique à la CIB a été synthétisée par la technologie Idaho, comme le montre la figure 1. La sonde oligo-nucléotidique FAM/Cy5.5/BHQ3 a été synthétisée à partir de l’extrémité 3′ avec un verre à pores contrôlé (CPG) non fluorescent Black Hole Quencher (BHQ-3). L’extrémité 5′ a ensuite été conjuguée avec une phosporamidite Cy5.5 suivie d’une phosporamidite terminale de fluorescéine 5′ (6-FAM).
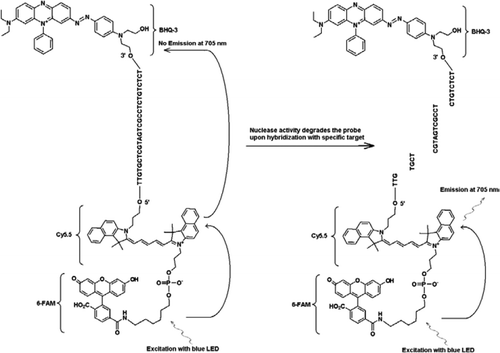
La sonde FRET-TaqMan contient trois étiquettes : un fragment d’extinction de trou noir à l’extrémité du bras 3′, un fluorophore émetteur (Cy5.5) et un fluorophore récolteur (FAM) réunis à l’extrémité de son bras 5′. Le FAM absorbe efficacement l’énergie de la LED bleue en tant que source de lumière. En l’absence de cibles, la sonde est sombre car l’énergie absorbée par le FAM est transférée à Cy5.5 et à son tour, l’énergie libérée par Cy5.5 est transféré à l’éteignoir et est perdu sous forme d’énergie thermique. En présence de cibles, la sonde TaqMan est clivée par l’activité de la Taq polymérase et l’inhibiteur est libéré et l’énergie absorbée par la FAM est transférée à Cy5.5 par le mécanisme de FRET pour émettre une fluorescence à 705 nm.
Analyse PCR en temps réel
Cytométrie en flux triée des oocystes de Cryptosporidium parvum (200 oocystes par tube) obtenus auprès du State Laboratory of Hygiene de l’Université du Wisconsin (Madison, WI, USA). ADN de C. les oocystes de parvum ont été extraits selon un protocole décrit par Hill et al. (12) et en suspension dans un tampon Tris EDTA (TE, pH 8,0) de 80 µL. Deux micro-litres d’ADN ont été ajoutés par réaction, ce qui correspond à 5 oocystes (équivalent à 100 copies d’ADN cible). Le mélange réactionnel d’amplification était constitué d’un mélange réactionnel de sonde de Quantifast PCR + kit ROX (Cat. no 204354; Qiagen) sans ROX ajouté, amorces JVAF, JVAR (0,4 µM chacune), sonde JVAP (0,2 µM), sonde JPIPC (0,2 µM) et 100 copies de la CIB. Une aliquote (2 µL) de l’échantillon d’ADN extrait a été ajoutée au capillaire réactionnel (Roche Diagnostics, Indianapolis, IN, USA) contenant 18 µL de mélange réactionnel. Un témoin positif de l’ADN génomique de C. parvum (Figure 2, graphique 1), le plasmide IPC (Figure 2, graphique 2), un mélange du témoin positif de C. parvum et du CIB (Figure 2, graphique 3), et un témoin sans matrice (NTC) constitué d’eau exempte de nucléase (Cat. aucun. AM9932; Ambion, Foster City, CALIFORNIE, États-Unis) au lieu de l’ADN (Figure 2, parcelle 4) ont été inclus dans chaque expérience. Le protocole a pris 30 minutes à compléter avec les conditions de PCR suivantes: étape de dénaturation à démarrage à chaud à 95° C pendant 3 min, suivie de 45 cycles avec une dénaturation à 95° C pendant 5 s, un recuit à 60° C pendant 30 s avec une seule acquisition de fluorescence (avec une vitesse de rampe de température de 20° C/s). Un résultat positif a été enregistré pour FAM dans le canal F1 (530 nm) et pour Cy5.5 dans le canal F3 (705 nm). Les réactions d’amplification ont été réalisées en trois exemplaires. Des amplicons (159 pb) ont été visualisés par électrophorèse sur gel d’agarose et coloration à l’acide nucléique vert brillant (eEnzyme, Gaithersburg, MD, USA) pour confirmer la spécificité du produit PCR.
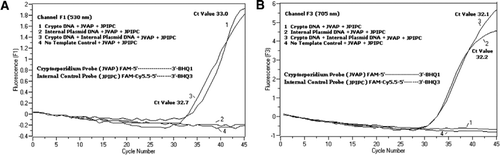
Un mélange équimolaire d’un FAM/BHQ1 et d’un FAM/Cy5.5/BHQ3 a été ajouté dans tous les mélanges réactionnels de PCR. (A) Le canal F1 surveille l’augmentation de la fluorescence FAM pour détecter la présence de C. parvum. La parcelle 1 (le canal F1 recueille principalement le signal FAM) contenant uniquement de l’ADN de C. parvum a révélé un signal FAM spécifique au Cryptosporidium dans la PCR. La parcelle 2 ne contenait que de l’ADN plasmidique et aucun signal FAM de la sonde FAM/Cy5.5/BHQ3. La parcelle 3 contenait les deux C. parvum et ADN plasmidique mais n’ont produit que le signal FAM spécifique à C. parvum. La parcelle 4 était un échantillon négatif contenant uniquement de l’eau exempte de nucléase à la place de tout ADN modèle; aucune fluorescence n’a été observée à partir des sondes. (B) Le canal F3 surveille l’augmentation de Cy5.5 pour la présence d’ADN plasmidique de contrôle interne. La parcelle 1 ne contenait que de l’ADN de C. parvum et aucun signal de fluorescence de la sonde FAM/BHQ1. La parcelle 2 (le canal F3 recueille principalement le signal Cy5.5) contenant uniquement du signal Cy5.5 spécifique au contrôle interne des plasmides (CIB) révélé par l’ADN plasmidique dans la PCR. La parcelle 3 contenait les deux C. parvum et ADN plasmidique mais n’ont produit que le signal Cy5.5 spécifique à la CIB. La parcelle 4 était un échantillon négatif contenant uniquement de l’eau exempte de nucléase à la place de tout ADN modèle; aucune fluorescence n’a été observée à partir des sondes.
Résultats et discussion
Des signaux de fluorescence PCR en temps réel reflétant l’amplification de l’ADN génomique de C. parvum ont été détectés dans le canal F1 (Figure 2A) en raison du marquage FAM de la sonde TaqMan. Une amplification réfléchissante du signal de fluorescence de la CIB a été détectée dans le canal F3 (Figure 2B) en raison de FAM/Cy5.5 étiquetage de la sonde IPC TaqMan. Cette sonde a produit une augmentation de Cy5.5 dans le canal F3 (Cy5.5), mais aucune augmentation de FAM dans le canal F1 (FAM) lorsque BHQ3 est séparé du FAM / Cy5.5 en raison de l’activité nucléase, car l’énergie a été transférée de FAM à Cy5.5. De même, dans la réaction duplex, l’amplification de l’ADN génomique de C. parvum a été détectée dans le canal F1, tandis que l’amplification IPC a été détectée simultanément dans le canal F3. FAM a une excitation à 495 nm et une émission à 520 nm. Cy5.5 a une excitation de 690 nm et une émission de 705 nm. Avec une telle proximité entre Cy5.5 et FAM sur la sonde IPC à triple marquage, le fluorophore Cy5.5 émet une fluorescence due au transfert complet d’énergie du fluorophore FAM, ce qui n’entraîne aucune émission de fluorescence de FAM dans le canal F1 (Figure 2A). Les réactions témoins négatives n’ont pas montré d’augmentation de la fluorescence due à FAM ou Cy5.5. Ces résultats démontrent également qu’une seule source LED bleue (470 nm) est suffisante pour surveiller les émissions de fluorescence double à travers les canaux F1 (530 nm) et F3 (705 nm) sans chevauchement spectral dans les canaux de surveillance respectifs pour FAM et Cy5.5. La technique de FRET-TaqMan a également été testée à l’aide d’un panel de 12 extraits d’ADN provenant d’échantillons de selles (8 provenant de spécimens précédemment déterminés comme positifs pour Cryptosporidium spp. et quatre spécimens négatifs pour Cryptosporidium spp.). L’analyse de l’ARNr 18S a permis de détecter les huit Cryptosporidium spp. Le test IPC TaqMan n’amplifie que l’ADN plasmidique IPC. Le test IPC a abouti à des valeurs de Ct de 32 à 37, ce qui indique jusqu’à 5 valeurs de Ct d’inhibition de la PCR pour certains échantillons de selles (32 était la valeur de Ct attendue pour les réactions de non-inhibition).
La présente technique diffère de la FRETTE traditionnellement utilisée en ce qu’une sonde oligonucléotidique marquée par fluorescence est utilisée (sonde FRET-TaqMan) au lieu de deux sondes (sondes FRETTE) dans lesquelles une sonde contient le fluorophore donneur et l’autre sonde contient le fluorophore accepteur (13,14). La sonde FRET-TaqMan est marquée à l’extrémité 5′ par deux fluorophores de fluorophores donneur (FAM) et accepteur (Cy5.5) et à l’extrémité 3′ par un extincteur BHQ-3 comme une sonde TaqMan. La région de liaison de sonde unique est sélectionnée pour se différencier de l’ADN cible de manière à surveiller deux mesures de fluorescence différentes dans deux canaux de détection différents (530 nm en F1 et 705 nm en F3) à partir de la même réaction d’amplification sans qu’il soit nécessaire d’inclure un logiciel de compensation de couleur. L’ADN cible et l’IPC avaient les mêmes sites de liaison de l’amorce mais une séquence de sonde différente. Cela garantit que l’utilisation des mêmes amorces pour les deux cibles d’ADN minimise la formation d’amorces-dimères ou l’interaction entre les ensembles d’amorces. Lorsque les fluorophores FAM / Cy5.5 sont placés de près à l’extrémité 5′, le transfert d’énergie de résonance a lieu avec une augmentation de l’émission de fluorescence du fluorophore Cy5.5. Cependant, l’utilisation de BHQ3, un accepteur de non fluorescence pour Cy5.5, diminue la fluorescence de fond potentielle, ce qui entraîne un rapport signal sur bruit plus élevé. Il a été constaté que l’addition de 100 copies de la CIB à une réaction contenant1100 copies de C génomique. l’ADN de parvum a donné des valeurs Ct similaires (Ct = 32) lorsque l’essai TaqMan de Cryptosporidium a été effectué en tant qu’essai singleplexe et en tant que test duplex avec l’essai TaqMan IPC. Lorsqu’elle a été appliquée à un panel d’échantillons d’ADN positifs au Cryptosporidium, la CIB a indiqué que certains échantillons ne contenaient pas d’inhibiteurs de PCR (sur la base des valeurs Ct attendues de 32), mais que d’autres échantillons étaient associés à une inhibition allant jusqu’à 5 valeurs Ct.
Le contrôle positif par PCR interne nouvellement développé s’est avéré sensible et spécifique pour la détection simultanée de deux cibles dans la même réaction en raison du marquage avec les FAM/ Cy5.5-BHQ3 et FAM / BHQ1 pour le gène cible respectif de l’ARNR IPC et 18S dans une PCR multiplex bicolore pour la surveillance dans les canaux de fluorescence F1 et F3 de l’instrument R.A.P.I.D. L’utilisation de la sonde étiquetée 5’FAM-Cy5.5/3’BHQ-3 comme second rapporteur est avantageuse car les maxima d’émission des deux sondes étaient bien espacés l’un de l’autre. La conception de la sonde à triple marquage FRET-TaqMan pourrait avoir des applications dans le développement d’un dispositif « laboratoire sur puce” jetable et peu coûteux avec une optique composée d’une excitation LED bleue et de deux sources d’émission de 530 nm et 705 nm, avec des filtres optiques appropriés pour détecter FAM dans un canal et Cy5.5 dans l’autre. Cette combinaison ne nécessiterait pas deux systèmes de source de fluorescence et de détection de haute puissance.
Remerciements
Les auteurs reconnaissent l’aide de Mike Powers au Centre de séquençage de l’ADN et de génomique (Université de l’Utah, Salt Lake City, UT, États-Unis) et de Rachelle Muller à Idaho Technology, Inc. pour discussion sur le développement du contrôle positif interne du cycleur R.A.P.I.D. Cette publication a été soutenue en partie par des fonds mis à disposition par l’intermédiaire des Centers for Disease Control and Prevention, du Bureau de coordination de la préparation au terrorisme et des interventions d’urgence. L’utilisation des noms commerciaux et des sources commerciales est uniquement à des fins d’identification et n’implique pas l’approbation des Centers for Disease Control and Prevention ou du Département américain de la Santé et des Services sociaux.
- 1. Il s’agit de l’un des plus importants ouvrages de l’histoire du Canada. 2005. Développement d’un nouveau contrôle positif interne pour les tests à base de Taqman. Mol. Cellule. Sondes 19:51-59.Crossref, Medline, CAS, Google Scholar
- 2. Il s’agit de l’un des principaux organismes de recherche et de recherche en sciences de la santé et en technologie de l’information. 2004. Considérations pratiques dans la conception de contrôles d’amplification internes pour les tests de PCR diagnostiques. J. Clin. Microbiol. 42:1863–1868.Crossref, Medline, CAS, Google Scholar
- 3. Betsou, F., K. Beaumont, J.M. Sueur et J. Orfila. 2003. Construction et évaluation de l’ADN de contrôle interne pour l’amplification par PCR de l’ADN de Chlamydia trachomatis à partir d’échantillons d’urine. J. Clin. Microbiol. 41:1274–1276.Crossref, Medline, CAS, Google Scholar
- 4. Burggraf, S. et B. Olgemoller. 2004. Technique simple pour le contrôle interne des tests d’amplification en temps réel. Clin. Chem. 50:819–825.Crossref, Medline, CAS, Google Scholar
- 5. Drsten, C., M. Weber, E. Seifried et W.K. Roth. 2000. Évaluation d’un nouveau test PCR avec séquence de contrôle interne compétitive pour le dépistage des donneurs de sang. Transfusion 40:718-724.Crossref, Medline, CAS, Google Scholar
- 6. Il s’agit d’une espèce de poissons de la famille des Pygmées. 2001. Quantification de l’ADN viral par PCR en temps réel en appliquant une amplification duplex, une standardisation interne et une détection de fluorescence bicolore. Appl. Environ. Microbiol. 67:2837–2839.Crossref, Medline, CAS, Google Scholar
- 7. Rosenstraus, M., Z. Il s’agit d’une espèce de poisson de la famille des Chang. 1998. Un contrôle interne pour la PCR diagnostique de routine: conception, propriétés et effet sur la performance clinique. J. Clin. Microbiol. 36:191–197.Medline, CAS, Google Scholar
- 8. Stacher, M., V. Leb et J. Berg. 2003. Une approche pratique pour la génération d’ADN de contrôle interne multiple pour un panel de tests PCR en temps réel. J. Virol. Méthodes 108:1-8.Crossref, Medline, CAS, Google Scholar
- 9. Zimmermann, K. et J.W. Mannhalter. 1996. Aspects techniques de la PCR compétitive quantitative. BioTechniques 21:268-279.Lien, CAS, Google Scholar
- 10. Jothikumar, N., A.J. da Silva, I. Moura, Y. Qvarnstrom et V.R. Hill. 2008. Détection et différenciation de Cryptosporidium hominis et Cryptosporidium parvum par double dosage de TaqMan. J. Med. Microbiol. 57:1099–1105.Les résultats de cette étude sont les suivants : Altschul, S.F., W. Gish, W. Miller, E.W. Myers et D.J. Lipman. 1990. Outil de recherche d’alignement local de base. J. Mol. Biol. 215:403–410.Les résultats de cette étude sont les suivants : Hill, V.R., A.m. Kahler, N. Jothikumar, T.B. Johnson, D. Hahn et T.L. Cromeans. 2007. Évaluation multiétatique d’une procédure basée sur l’ultrafiltration pour la récupération simultanée de microbes entériques dans des échantillons d’eau du robinet de 100 litres. Appl. Environ. Microbiol. 73:4218–4225.Les résultats de cette étude sont les suivants : Bernard, P.s., M.J. Lay et C.T. Wittwer. 1998. Amplification et détection intégrées de la mutation ponctuelle C677T dans le gène de la méthylènetétrahydrofolate réductase par transfert d’énergie par résonance de fluorescence et courbes de fusion de la sonde. Anal. Biochem. 255:101–107.Les résultats de cette étude sont les suivants : Wittwer, C.T., M.G. Herrmann, A.A. Moss et R.P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130–138.Link, CAS, Google Scholar