Diseño de sondas FRET-TaqMan para PCR multiplex en tiempo real utilizando un control positivo interno
Introducción
La PCR en tiempo real permite la monitorización continua de la fluorescencia de fluoróforos durante la generación de productos de PCR en un formato de tubo cerrado. Actualmente, los métodos disponibles utilizan sondas etiquetadas o tinte intercalado de ADN para monitorear la amplificación del producto de PCR. La sonda TaqMan es un oligonucleótido monocatenario que contiene un fluoróforo y un apagador separados entre 10 y 30 bases. La sonda TaqMan se hidroliza durante la amplificación debido a la actividad de exonucleasa específica de doble hebra 5′-3′ de la polimerasa Taq que separa el fluoróforo del apagador, lo que resulta en un aumento de fluorescencia. Los instrumentos de PCR en tiempo real están equipados con detectores de fluorescencia y software capaz de estimar el umbral de ciclo (Ct), el ciclo en el que la fluorescencia es mayor que la fluorescencia de fondo, para reacciones positivas. Sin embargo, la técnica no puede diferenciar un resultado negativo verdadero de un falso negativo cuando la PCR se ve afectada por inhibidores de amplificación. Después de la extracción de ácido nucleico, se informa que los inhibidores pueden estar presentes en muestras clínicas (por ejemplo, hemoglobina), muestras ambientales (por ejemplo, ácidos húmicos y fúlvicos) y productos químicos empleados durante la extracción de ácido nucleico (por ejemplo, etanol, detergentes o agentes chaotrópicos). La fiabilidad de los ensayos de diagnóstico aumenta con la inclusión de un ácido nucleico de control interno que puede indicar la presencia e impacto de inhibidores de la PCR (1,2). Un control positivo interno (IPC) se amplifica simultáneamente en presencia de una secuencia objetivo utilizando un fluoróforo etiquetado que emite luz a una longitud de onda diferente al fluoróforo utilizado para el ensayo de secuencia objetivo, con los dos fluoróforos detectados en diferentes canales por el instrumento de PCR en tiempo real. El control interno comúnmente utilizado para la PCR es un plásmido que contiene una secuencia similar a la del objetivo del ensayo, excepto para la región de la sonda. Se añade un número limitado de moléculas de CIP al objetivo de ensayo individual y se amplifica conjuntamente con el ácido nucleico objetivo; por lo tanto, una señal IPC positiva es evidencia de que la reacción de amplificación procedió lo suficiente para generar una señal positiva a partir de cantidades muy pequeñas de ácido nucleico objetivo. Esta característica es importante para garantizar una amplificación equivalente del CIP y del ácido nucleico objetivo (3-9). Sin embargo, algunos dispositivos, como el LightCycler 1.2 y el LightCycler 2 (Roche Applied Science, Indianápolis, IN, EE.) instrumentos (Idaho Technology, Salt Lake City, UT, EE. UU.) e instrumentos portátiles de PCR en tiempo real: solo están equipados con una fuente de luz y un canal de emisión asociado. La multiplexación en este tipo de instrumentos requiere el uso de oligonucleótidos multicelulares y el concepto de transferencia de energía de resonancia de fluorescencia (FRET). Sin embargo, las sondas de TRASTE pueden ser difíciles de diseñar y pueden carecer de robustez en regiones hipervariables y cuando están presentes condiciones de amplificación no óptimas. El uso de sondas de TRASTE requiere la identificación de dos regiones para dos sondas. El sistema de sonda FRET se basa en la hibridación adyacente de oligonucleótidos de las dos sondas de una sola etiqueta de tal manera que la fluorescencia del tinte aceptor se detecta a través de la excitación del fluoróforo unido a la segunda sonda. Una desventaja importante de la sonda de TRASTE es el requisito de un área de secuencia más grande necesaria para acomodar dos sondas adyacentes con un espacio de 2-5 nucleótidos. Existe la posibilidad de multiplexar con otros sistemas de etiquetas, como MegaStokes dye (DYXL; Dyomics, Jena, Alemania) y Pulsar 650 (Biosearch Technologies, Inc., Novato, CA, EE. UU.), pero estos sistemas de etiquetado requieren información espectral para compensar la diafonía del canal y no están ampliamente disponibles. La sonda FRET-TaqMan desarrollada en este estudio, sin embargo, no requiere software de compensación de color y utiliza fluoróforos (FAM, Cy5.5) que están ampliamente disponibles. El presente informe se centra en el diseño de una sonda de triple etiqueta para permitir la multiplexación en instrumentos equipados con una sola fuente de excitación de diodo emisor de luz azul (LED) (470 nm) y los tres canales de detección correspondientes asociados (por ejemplo, 530 nm, 640 nm y 705 nm).
Materiales y métodos
Principio de diseño de la sonda
Todos los experimentos se realizaron en el sistema R. A. P. I. D. de Idaho Technology, un instrumento portátil de PCR en tiempo real de 32 muestras equipado con un led azul para excitación (470 nm) y tres canales de detección de fluorescencia asociados que miden la fluorescencia a 530, 640 y 705 nm para ensayos monocromáticos y de doble color. Los cebadores JVAF y JVAR (10) y la sonda 5′-FAM/BHQ-1 se sintetizaron en las Instalaciones Centrales de Biotecnología de los CDC (Atlanta, GA, EE.UU.) y la sonda 5’FAM-Cy5. 5/3 ‘ BHQ-3 se sintetizó en Idaho Technology. Para habilitar la PCR TaqMan dúplex, se etiquetó una sonda con 5’-FAM/BHQ-1 (longitud de onda de fluorescencia de 520 nm) y otra con 5’FAM-Cy5.5/3 ‘ BHQ-3 (longitud de onda de fluorescencia de 705 nm). En solución, la sonda etiquetada con 5’FAM-Cy5.5/3’BHQ-3 permanece apagada al hibridarse a una secuencia, y después de la división posterior de la sonda por extensión de imprimación enzimática, se elimina el apagador. Esto resulta en el TRASTE a través de la excitación de FAM por la fuente de luz del instrumento R. A. P. I. D. La energía de excitación se transfiere al fluoróforo receptor, Cy5.5, y la fluorescencia emitida se mide en un segundo canal de fluorescencia (canal de fluorescencia F3 para el instrumento R. A. P. I. D.). La emisión de fluorescencia Cy5.5 es el resultado de la combinación de técnicas de sonda de TRASTE y TaqMan, por lo que estas sondas de triple etiqueta pueden denominarse sondas de «TRASTE-TaqMan».
Cebadores, sondas y plásmidos
Para demostrar las capacidades de multiplexación del diseño de FRET-TaqMan, utilizamos un ensayo de PCR en tiempo real de Cryptosporidium específico de género para amplificar una porción de 159 bp del gen 18S rRNA (10). La imprimación delantera (JVAF) era de 5′-ATGACGGGTAACGGGAAT-3′, la imprimación inversa (JVAR) era de 5′-CCAATTACAAAAC-CAAAAAGTCC-3′, y la sonda TaqMan (JVAP) era de 5′-FAM-CGCGCCTGCT-GCCTTCCTTAGATG-BHQ1–3′, correspondiente a los nucleótidos 100-118, 258-236, y 184 a 161, respectivamente, de la adhesión al GenBank no. AY458612. Los cebadores para la construcción del Cryptosporidium IPC contenían el mismo conjunto de secuencias de cebadores reportadas para la amplificación de Cryptosporidium spp., a excepción de la región de unión de la sonda del ADN objetivo, que fue reemplazada por la secuencia artificial 5′-TTGTGCTCGTAGTCGCCTCTGTCTCT-3′ (JPIPC). Esta secuencia de sonda tenía una temperatura de recocido de 69°C y fue diseñada para no tener estructura secundaria. La especificidad teórica para la sonda interna se determinó mediante una búsqueda EXPLOSIVA (11) de las secuencias de sonda y cebador en las que no se encontró coincidencia exacta con ninguna secuencia de criptosporidio. El fragmento de CIP fue sintetizado y clonado en un plásmido mediante Tecnologías Integradas de ADN (Coralville, IA, EE. UU.). Se añadió una concentración conocida de ADN plásmido (100 copias/reacción) a la mezcla maestra de todas las reacciones, excepto en el control negativo. Una sonda de triple etiqueta (JPIPC) específica para el IPC fue sintetizada por Tecnología de Idaho, como se muestra en la Figura 1. La sonda de oligonucleótido FAM/Cy5.5/BHQ3 se sintetizó a partir del extremo 3′ con vidrio de poros controlado (CPG) con apagador de agujeros negros no fluorescente (BHQ-3). El extremo de 5′ se conjugó con una fosforamidita Cy5.5 seguida de una fosforamidita terminal de 5′ fluoresceína (6-FAM).
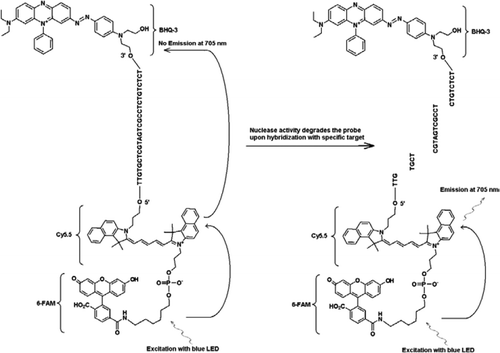
La sonda FRET-TaqMan contiene tres etiquetas: una mitad apagadora de agujeros negros al final del brazo de 3′, un fluoróforo emisor (Cy5.5) y un fluoróforo cosechador (FAM) unidos al final de su brazo de 5′. La FAM absorbe de manera eficiente la energía del LED azul como fuente de luz. En ausencia de objetivos, la sonda está oscura porque la energía absorbida por la FAM se transfiere a Cy5.5 y, a su vez, la energía liberada de Cy5.5 se transfiere al apagador y se pierde como energía térmica. En presencia de objetivos, la sonda TaqMan es escindida por la actividad de la polimerasa Taq y se libera un apagador y la energía absorbida por la FAM se transfiere a Cy5.5 a través del mecanismo de TRASTE para emitir fluorescencia a 705 nm.
Ensayo de PCR en tiempo real
Ooquistes de Cryptosporidium parvum clasificados por citometría de flujo (200 ooquistes por tubo) obtenidos del Laboratorio Estatal de Higiene de la Universidad de Wisconsin (Madison, WI, EE. ADN de C. los ooquistes de parvum se extrajeron utilizando un protocolo descrito por Hill et al. (12) y suspendido en tampón Tris EDTA (TE, pH 8,0) de 80 µL. Se agregaron dos micro litros de ADN por reacción, que correspondieron a 5 ooquistes (equivalentes a 100 copias de ADN diana). La mezcla de reacción de amplificación consistió en una mezcla de reacción del kit PCR + ROX de sonda cuantificada (Cat. no. 204354; Qiagen) sin ROX añadido, JVAF, cebadores JVAR (0,4 µM cada uno), sonda JVAP (0,2 µM), sonda JPIPC (0,2 µM) y 100 copias del IPC. Se añadió una alícuota (2 µL) de la muestra de ADN extraída al capilar de reacción (Roche Diagnostics, Indianápolis, IN, EE.UU.) que contenía una mezcla de reacción de 18 µL. Un control positivo de ADN genómico de C. parvum (Figura 2, gráfico 1), el plásmido IPC (Figura 2, gráfico 2), una mezcla del control positivo de C. parvum y el IPC (Figura 2, gráfico 3), y un control sin plantilla (NTC) que consiste en agua libre de nucleasas (Cat. no. AM9932; Ambion, Foster City, CA, EE. UU.) en lugar del ADN (Figura 2, gráfico 4) se incluyeron en cada experimento. El protocolo tardó 3 30 minutos en completarse con las siguientes condiciones de PCR: etapa de desnaturalización de arranque en caliente a 95°C durante 3 min, seguida de 45 ciclos con desnaturalización de 95 ° C durante 5 s, recocido de 60°C durante 30 s con adquisición de fluorescencia única (con una tasa de rampa de temperatura de 20°C/s). Se registró un resultado positivo para FAM en el canal F1 (530 nm) y para Cy5, 5 en el canal F3 (705 nm). Las reacciones de amplificación se llevaron a cabo por triplicado. Los amplicones (159 pb) se visualizaron mediante electroforesis en gel de agarosa y tinción de ácido nucleico verde brillante (eEnzyme, Gaithersburg, MD, EE.UU.) para confirmar la especificidad del producto de PCR.
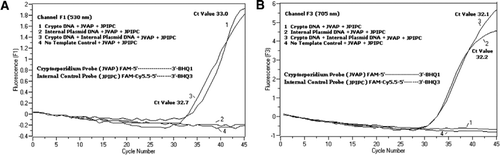
Se añadió una mezcla equimolar de un FAM/BHQ1 y un FAM/Cy5, 5/BHQ3 en todas las mezclas de reacción PCR. A) Los monitores de canal F1 aumentan la fluorescencia FAM para detectar la presencia de C. parvum. Gráfico 1 (el canal F1 recoge principalmente la señal FAM) que contiene solo ADN de C. parvum que reveló la señal FAM específica de Criptosporidio en la PCR. La gráfica 2 contenía solo ADN plásmido y ninguna señal FAM de la sonda FAM/Cy5.5/BHQ3. La parcela 3 contenía ambos C. adn de parvum y plásmido, pero solo produjo señal FAM específica de C. parvum. La gráfica 4 era una muestra negativa que contenía solo agua libre de nucleasas en lugar de cualquier ADN de plantilla; no se observó fluorescencia de las sondas. B) Aumento de los monitores de canal F3 en Cy5. 5 para detectar la presencia de ADN plásmido de control interno. La gráfica 1 contenía solo ADN de C. parvum y ninguna señal de fluorescencia de la sonda FAM / BHQ1. Gráfico 2 (el canal F3 recoge principalmente la señal Cy5.5) que contiene solo la señal Cy5.5 específica del control interno de plásmidos revelada por ADN plásmido (IPC) en la PCR. La parcela 3 contenía ambos C. adn de parvum y plásmido, pero solo produjo señal CY5.5 específica de IPC. La gráfica 4 era una muestra negativa que contenía solo agua libre de nucleasas en lugar de cualquier ADN patrón; no se observó fluorescencia de las sondas.
Resultados y discusión
Señales de fluorescencia de PCR en tiempo real que reflejan la amplificación del ADN genómico de C. parvum se detectó en el canal F1 (Figura 2A) debido al etiquetado FAM de la sonda TaqMan. Se detectó amplificación reflectante de señales de fluorescencia del CIP en el canal F3 (Figura 2B) debido a FAM/Cy5.5 etiquetado de la sonda IPC TaqMan. Esta sonda produjo un aumento de Cy5.5 en el canal F3 (Cy5.5), pero no un aumento de FAM en el canal F1 (FAM) cuando BHQ3 se separa del FAM/Cy5.5 debido a la actividad de la nucleasa, porque la energía se transfirió de FAM a Cy5.5. De manera similar, en la reacción dúplex, se detectó la amplificación genómica de ADN de C. parvum en el canal F1, mientras que la amplificación IPC se detectó simultáneamente en el canal F3. FAM tiene una excitación a 495 nm y una emisión a 520 nm. Cy5. 5 tiene una excitación de 690 nm y una emisión de 705 nm. Con tal proximidad entre Cy5.5 y FAM en la sonda IPC de triple etiqueta, el fluoróforo Cy5.5 emite fluorescencia debido a la transferencia completa de energía desde el fluoróforo FAM, lo que resulta en una emisión de fluorescencia de FAM en el canal F1 (Figura 2A). Las reacciones de control negativo no mostraron un aumento de fluorescencia debido a FAM o Cy5.5. Estos resultados también demuestran que una única fuente de LED azul (470 nm) es suficiente para monitorear las emisiones de fluorescencia dual a través de los canales F1 (530 nm) y F3 (705 nm) sin superposición espectral en los respectivos canales de monitoreo para FAM y Cy5.5. La técnica de FRET-TaqMan también se probó utilizando un panel de 12 extractos de ADN de muestras de heces (8 de muestras previamente determinadas como positivas para Cryptosporidium spp. y cuatro especímenes que fueron negativos para Cryptosporidium spp.). El ensayo 18S rRNA fue capaz de detectar los ocho Cryptosporidium spp. El ensayo IPC TaqMan sólo amplifica el ADN plásmido de IPC. El ensayo IPC dio como resultado valores de Tc de 32-37, que indicaron hasta 5 valores de Tc de inhibición de la PCR para algunas muestras de heces (32 fue el valor esperado de Tc para reacciones sin inhibición).
La presente técnica difiere de la utilizada tradicionalmente en que se utiliza una sonda de oligonucleótidos etiquetada fluorescentemente (sonda FRET-TaqMan) en lugar de dos sondas (sondas FRET) en las que una sonda contiene el fluoróforo donante y la otra sonda contiene el fluoróforo receptor (13,14). La sonda FRET-TaqMan está etiquetada en el extremo de 5′ con dos fluoróforos de donante (FAM) y receptor (Cy5.5) y en el extremo de 3′ con un apagador BHQ-3 como una sonda TaqMan. La región de unión de sonda única se selecciona para diferenciarse del ADN objetivo a fin de monitorear dos mediciones de fluorescencia diferentes en dos canales de detección diferentes (530 nm en F1 y 705 nm en F3) de la misma reacción de amplificación sin la necesidad de incluir software de compensación de color. El ADN objetivo y el CIP tenían los mismos sitios de unión de cebador pero una secuencia de sonda diferente. Esto garantiza que el uso de los mismos cebadores para ambos objetivos de ADN minimice la formación de dímeros o la interacción entre los conjuntos de cebadores. Cuando los fluoróforos FAM / Cy5. 5 se colocan de cerca en el extremo de 5′, la transferencia de energía de resonancia tiene lugar con un aumento en la emisión de fluorescencia del fluoróforo Cy5.5. Sin embargo, el uso de BHQ3, un aceptor de no fluorescencia para Cy5.5, disminuye la fluorescencia potencial de fondo, lo que resulta en una relación señal-ruido más alta. Se encontró que la adición de 100 ejemplares de la CIP para una reacción que contiene ∼100 copias de genómica C. el ADN parvum dio como resultado valores de Tc similares (Ct = 32) cuando el ensayo Cryptosporidium TaqMan se realizó como un ensayo de un solo complejo y como un ensayo dúplex con el ensayo IPC TaqMan. Cuando se aplicó a un panel de muestras de ADN con Criptosporidio positivo, el IPC indicó que algunas muestras no contenían inhibidores de PCR (en base a los valores esperados de Tc de 32), pero otras muestras se asociaron con inhibición de hasta 5 valores de Tc.
El control positivo de PCR interno recientemente desarrollado demostró ser sensible y específico para la detección simultánea de dos blancos en la misma reacción debido al etiquetado con el FAM/Cy5.5-BHQ3 y FAM/BHQ1 para el respectivo gen diana IPC y 18S rRNA en una PCR múltiple de doble color para monitoreo en canales de fluorescencia F1 y F3 del instrumento R. A. P. I. D. El uso de la sonda etiquetada con 5’FAM-Cy5.5/3’BHQ-3 como segundo reportero es ventajoso porque los máximos de emisión de las dos sondas estaban bien espaciados entre sí. El diseño de sonda de triple etiqueta FRET-TaqMan podría tener aplicaciones en el desarrollo de un dispositivo desechable de bajo costo «laboratorio en un chip» con óptica que consta de un led azul de excitación y dos fuentes de emisión de 530 nm y 705 nm, con filtros ópticos adecuados para detectar FAM en un canal y Cy5.5 en el otro. Esta combinación no requeriría dos sistemas de detección y fuente de fluorescencia de alta potencia.
Agradecimientos
Los autores agradecen la asistencia de Mike Powers en la Instalación de Secuenciación de ADN y Núcleo Genómico (Universidad de Utah, Salt Lake City, UT, EE.UU.) y Rachelle Muller en Idaho Technology, Inc. para la discusión sobre el desarrollo del control positivo interno para el ciclador R. A. P. I. D. Esta publicación se financió en parte con fondos aportados a través de los Centros para el Control y la Prevención de Enfermedades, la Oficina de Coordinación de la Preparación para el Terrorismo y la Respuesta de Emergencia. El uso de nombres comerciales y fuentes comerciales es solo para identificación y no implica aprobación por parte de los Centros para el Control y la Prevención de Enfermedades o el Departamento de Salud y Servicios Humanos de los Estados Unidos.
- 1. Hartman, L. J., S. R. Coyne, and D. A. Norwood. 2005. Desarrollo de un nuevo control positivo interno para ensayos basados en Taqman. Mol. Celular. Sondas 19: 51-59.Crossref, Medline, CAS, Google Scholar
- 2. Hoorfar, J., B. Malorny, A. Abdulmawjood, N. Cook, M. Wagner, and P. Fach. 2004. Consideraciones prácticas en el diseño de controles de amplificación internos para ensayos de PCR de diagnóstico. J. Clin. Microbiol. 42:1863–1868.Crossref, Medline, CAS, Google Scholar
- 3. Betsou, F., K. Beaumont, J. M. Sueur, and J. Orfila. 2003. Construction and evaluation of internal control DNA for PCR amplification of Chlamydia trachomatis DNA from urine samples. J. Clin. Microbiol. 41:1274–1276.Crossref, Medline, CAS, Google Scholar
- 4. Burggraf, S. y B. Olgemoller. 2004. Técnica sencilla para el control interno de ensayos de amplificación en tiempo real. Clin. Chem. 50:819–825.Crossref, Medline, CAS, Google Scholar
- 5. Drosten, C., M. Weber, E. Seifried, and W. K. Roth. 2000. Evaluación de un nuevo ensayo de PCR con secuencia de control interno competitiva para el cribado de donantes de sangre. Transfusion 40: 718-724.Crossref, Medline, CAS, Google Scholar
- 6. Gruber, F., F. G. Falkner, F. Dorner, and T. Hammerle. 2001. Cuantificación del ADN viral por PCR en tiempo real aplicando amplificación dúplex, estandarización interna y detección de fluorescencia de dos colores. Appl. Environ. Microbiol. 67:2837–2839.Crossref, Medline, CAS, Google Scholar
- 7. Rosenstraus, M., Z. Wang, S. Y. Chang, D. DeBonville, and J. P. Spadoro. 1998. Un control interno para PCR de diagnóstico rutinario: diseño, propiedades y efecto en el rendimiento clínico. J. Clin. Microbiol. 36:191–197.Medline, CAS, Google Scholar
- 8. Stocher, M., V. Leb, and J. Berg. 2003. Un enfoque conveniente para la generación de ADN de control interno múltiple para un panel de ensayos de PCR en tiempo real. J. Virol. Methods 108: 1-8.Crossref, Medline, CAS, Google Scholar
- 9. Zimmermann, K. y J. W. Mannhalter. 1996. Aspectos técnicos de la PCR competitiva cuantitativa. BioTechniques 21: 268-279.Link, CAS, Google Scholar
- 10. Jothikumar, N., A. J. da Silva, I. Moura, Y. Qvarnstrom, and V. R. Hill. 2008. Detección y diferenciación de Cryptosporidium hominis y Cryptosporidium parvum mediante ensayos duales de TaqMan. J. Med. Microbiol. 57:1099–1105.Crossref, Medline, CAS, Google Scholar
- 11. Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410.Crossref, Medline, CAS, Google Scholar
- 12. Hill, V. R., A. M. Kahler, N. Jothikumar, T. B. Johnson, D. Hahn, and T. L. Cromeans. 2007. Evaluación multiestatal de un procedimiento basado en la ultrafiltración para la recuperación simultánea de microbios entéricos en muestras de agua del grifo de 100 litros. Appl. Environ. Microbiol. 73:4218–4225.Crossref, Medline, CAS, Google Scholar
- 13. Bernard, P. S., M. J. Lay, and C. T. Wittwer. 1998. Amplificación y detección integradas de la mutación puntual C677T en el gen de la metilentetrahidrofolato reductasa mediante transferencia de energía de resonancia de fluorescencia y curvas de fusión de sonda. Anal. Bioquímica. 255:101–107.Crossref, Medline, CAS, Google Scholar
- 14. Wittwer, C. T., M. G. Herrmann, A. A. Moss, and R. P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130–138.Link, CAS, Google Scholar