Debilidad en las rodillas: Síndrome de Miller Fisher
El síndrome de Miller Fisher (MFS), una variante rara y autolimitada del síndrome de Guillain-Barré (SGB), es un síndrome de anticuerpos anti-GQ1bIgG que afecta a los sistemas nervioso central y periférico.1 Una afección polineuropática neuromuscular aguda, la MFS causa una parálisis ascendente con una tríada de presentación clásica de oftalmoplejía, ataxia y areflexia. Los investigadores describieron por primera vez la tríada clínica en 1932.1 Dos décadas más tarde, el especialista canadiense en accidentes cerebrovasculares Charles Miller Fisher fue el primero en publicar un informe en 1956 que describía los hallazgos clínicos y definía la afección como una forma limitada de EGB.2
Los síntomas ocurren durante varios días, a menudo en invierno y primavera, típicamente después de una infección viral, y los pacientes a menudo experimentan diplopía como uno de los primeros síntomas. Cuando un paciente presenta síntomas como esotropía intermitente, dificultades horizontales y pupilas midriáticas fijas, y su examen neurológico es anormal, es hora de pensar en EGB, específicamente en MFS. El siguiente caso ilustra los signos y síntomas relevantes y refleja el resultado típico.
Antecedentes
Un varón caucásico de 27 años de edad se presentó en la Clínica Oftalmológica del VA durante los meses de primavera, reportando diplopía binocular de inicio agudo que comenzó un día antes. El paciente afirmó que era constante, pero que solo ocurría a distancia con objetos desplazados horizontalmente. No hubo dolor ocular ni visión borrosa asociada con la diplopía. He also complained of recent dizziness and balance difficulties. Informó de una infección sinusal con congestión paranasal significativa que comenzó una semana antes de la visión doble. Los antecedentes oculares y médicos anteriores no eran notables; el paciente no estaba tomando ningún medicamento y no tenía alergias a ningún medicamento.
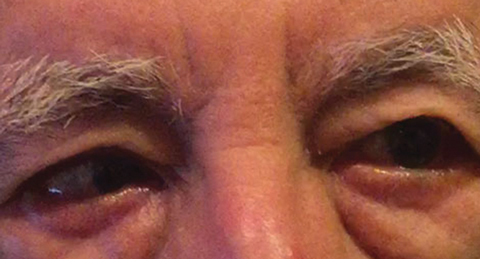
Fig. 1. El diagnóstico diferencial posterior al examen basal incluyó una parálisis parcial de la CN VI derecha, lo que podría indicar una condición vascular, como se observó aquí en otro paciente. Haga clic en la imagen para ampliarla. Foto: Michael DelGiodice, DO
Datos de diagnóstico
En el examen basal, las acuidades visuales no corregidas fueron 20/20-2 DO y 20/25-2 SG. Las pupilas eran de 5 mm igualmente redondas y reactivas a la luz sin defecto pupilar aferente. Las motilidades extraoculares eran completas, lisas y precisas en ambos ojos sin restricciones. La prueba de cobertura a distancia reveló una esotropía derecha intermitente de cuatro a seis dioptrías prismáticas en la mirada primaria que ocurrió aproximadamente el 50% del tiempo. La realización de la prueba de cobertura en near reveló una esoforia de dioptrías de cuatro prisma en la mirada primaria sin tropia.
Aunque no se realice en este caso, los médicos deben cubrir la prueba en la mirada primaria, derecha e izquierda. En pacientes con diplopía, la parálisis del nervio craneal seis (CN VI) se manifestará como desviación no compulsiva, peor en la mirada derecha o izquierda, dependiendo del lado en el que se encuentre la parálisis. Una foria descompensante, por otro lado, mostrará una desviación de comité.
El error de refracción fue de -0,50 D DE la esfera y -0,75 D de la esfera y la visión fue corregible a 20/20 en cada ojo. Las presiones intraoculares estaban dentro de los límites normales en ambos ojos. Los hallazgos de los segmentos anterior y posterior no fueron notables en los ojos derecho e izquierdo. Los nervios ópticos parecían sanos con una relación copa-disco de 0,15 en ambos ojos.
Los diagnósticos diferenciales incluyeron parálisis parcial de CN VI derecha (Figura 1), miastenia gravis, un proceso de compresión y una esoforia a distancia descompensante que causó una esotropía derecha intermitente. El paciente negó tener antecedentes de diplopía binocular.
La clínica oftalmológica ordenó imágenes del cerebro y las órbitas para descartar compromiso cerebral, incluida una lesión compresiva. El neurorradiólogo del personal realizó e interpretó exploraciones de tomografía computarizada (TC) del cerebro y las órbitas. Se eligió la TC en lugar de la resonancia magnética (RM) debido al tiempo y la disponibilidad; las pruebas y los resultados se podían obtener el mismo día con la TC en la clínica fuera de horario. Los resultados no fueron notables, sin signos de hemorragia intracraneal, lesión compresiva o infarto agudo.
La TC de los senos paranasales reveló enfermedad crónica del seno paranasal de moderada a grave y opacificación completa del seno maxilar derecho. No hubo extensión en la órbita. El médico de familia del paciente fue informado de los síntomas del paciente, los hallazgos del examen ocular y los resultados de la tomografía computarizada y comenzó el tratamiento para la sinusitis maxilar con un paquete de dosis de azitromicina oral para cinco días e instruyó al paciente para que realizara un seguimiento en la clínica oftalmológica.
El paciente regresó cinco días después de completar su ciclo de azitromicina oral y afirmó que no experimentó mejoría en la diplopía a distancia; además, experimentó la aparición de fotofobia significativa y dolor periocular alrededor de ambos ojos. El paciente afirmó que su desequilibrio en la marcha y las dificultades para caminar también estaban empeorando. En el examen, las agudezas visuales mejor corregidas permanecieron 20/20 en los ojos derecho e izquierdo. Las pupilas se dilataron a 8,5 mm sin reactividad a la luz en ninguno de los ojos (Figura 2). Las motilidades extraoculares revelaron restricción lateral con dolor al moverse en el ojo derecho y restricción medial con dolor al moverse en el ojo izquierdo consistente con parálisis de la mirada derecha (Figura 3). Las persecuciones horizontales y verticales eran sacádicas con movimientos inexactos y espasmódicos. Los hallazgos de los segmentos anterior y posterior se mantuvieron sin complicaciones.
El diagnóstico diferencial después del examen oftalmológico incluyó oftalmoplejía internuclear izquierda, parálisis de la mirada derecha y parálisis de la mirada derecha con oftalmoplejía internuclear izquierda, lo que podría explicar los hallazgos de motilidad extraocular. Otros diferenciales incluyen una lesión del mesencéfalo o miastenia grave. Los diagnósticos diferenciales de dilatación pupilar incluyen parálisis del tercer nervio (CN III), trauma y dilatación farmacológica. El paciente no presentó ningún otro hallazgo compatible con parálisis del tercer nervio y negó haber sufrido traumatismo o exposición a agentes farmacológicos.
El paciente fue remitido a neurología para su posterior evaluación. El examen neurológico informó que el paciente estaba completamente alerta y orientado sin pérdida de sensibilidad facial. El paciente presentó ataxia leve con dificultad para caminar en tándem y pararse en cuclillas. Se presentaron debilidad proximal en las extremidades superiores e inferiores y reflejos anormales. Todas las modalidades sensoriales estaban intactas. La resonancia magnética y la angiografía por resonancia magnética (ARM) del cerebro y las órbitas no fueron notables. La proteína del líquido cefalorraquídeo se elevó a 75 mg / dL (rango de referencia: 15 mg/dL a 45 mg/dL) sin otras anomalías. La prueba de electromiografía no fue notable y no mostró anomalías en la conducción nerviosa desde la columna vertebral hasta los pies y las manos.
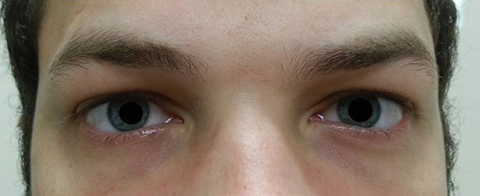
Fig. 2. Las pupilas del paciente estaban dilatadas 8,5 mm sin reactividad en los ojos derecho o izquierdo. Haga clic en la imagen para ampliarla.
Diagnóstico
El paciente presentó síntomas agudos de diplopía, ataxia y areflexia. Dada la esotropía derecha intermitente, las actividades horizontales deterioradas, la pupila midriática fija, la ataxia leve y los reflejos anormales en el examen neurológico y la proteína elevada del líquido cefalorraquídeo, se realizó el diagnóstico de MFS. El paciente fue ingresado en el hospital para vigilar cualquier otra complicación. Se proporcionó un parche ocular al paciente para cubrir el ojo derecho según fuera necesario para la diplopía.
Actualmente no existen criterios diagnósticos publicados para el síndrome de Miller Fisher. El diagnóstico generalmente se realiza a través de la presentación de la tríada clínica junto con estudios de imágenes y del líquido cefalorraquídeo. La resonancia magnética no suele ser notable en la afección, aunque algunos estudios han mostrado lesiones centrales en el mesencéfalo, el puente y la médula inferior. Los estudios electrofisiológicos muestran una conducción sensorial periférica reducida o anormal.17
La proteína del líquido cefalorraquídeo a menudo está elevada sin otros hallazgos anormales. En un estudio histórico, el 64,4% de los pacientes con MFS mostraron proteínas elevadas en el líquido cefalorraquídeo.8 Una prueba positiva de anticuerpos anti-GQ1b IgG permite un diagnóstico definitivo. En un estudio, el anticuerpo IgG anti-GQ1b estaba presente en hasta el 95% de los pacientes con la enfermedad y estaba ausente en los controles.3
El neurólogo diagnosticó al paciente con MFS basándose en la tríada clínica de oftalmoplejía, ataxia y areflexia, una resonancia magnética y una ARM del cerebro sin complicaciones y una proteína elevada del líquido cefalorraquídeo. Por lo tanto, en este caso no se realizó la prueba de anticuerpos anti-GQ1b IgG.
En el síndrome de Guillain-Barré y en la encefalitis del tronco encefálico de Bickerstaff también se observan varias características clínicas de la MFS, lo que dificulta el diagnóstico. Los pacientes con solo Guillain-Barré presentan debilidad en las extremidades, pérdida sensorial, neuropatía craneal y areflexia sin manifestaciones oftalmológicas. Los pacientes con encefalitis del tronco encefálico de Bickerstaff suelen presentar oftalmoplejía, ataxia, hiperreflexia y trastornos de la conciencia. Dado que hay varios síntomas y signos presentes en las tres afecciones, la prueba de títulos de anticuerpos IgG anti-GQ1b es útil para un diagnóstico más definitivo. El anticuerpo IgG anti-GQ1b es positivo en una gran mayoría de los pacientes con síndrome de Miller Fisher en comparación con el 66% en la encefalitis del tronco encefálico de Bickerstaff y el 26% de los pacientes con síndrome de Guillain-Barré.16
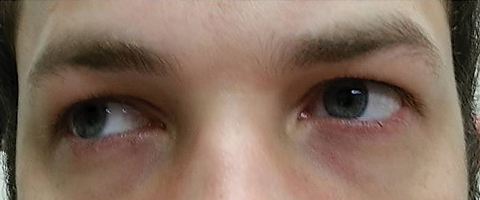
Fig. 3. Las motilidades extraoculares revelaron restricción en la dextroversión. Haga clic en la imagen para ampliarla.
Tratamiento y seguimiento
El paciente fue evaluado el día después del ingreso, cuando declaró que no experimentaba cambios visuales ni mejoría en el equilibrio. La diplopía binocular seguía siendo constante; por lo tanto, el ojo derecho permaneció parcheado. El dolor ocular con miradas lejanas y movimientos oculares rápidos todavía estaban presentes. En el examen de cabecera limitado, las agudezas visuales mejor corregidas permanecieron 20/20 en los ojos derecho e izquierdo. Pupilas dilatadas a las 8.0 mm en ambos ojos con reactividad mínima, que mejoró ligeramente desde el día anterior. La visión del color era normal en los ojos derecho e izquierdo. Las motilidades extraoculares estaban llenas en todos los cuadrantes con dolor aún presente en las miradas horizontales lejanas. Las búsquedas horizontales siguieron siendo sacádicas con movimientos irregulares y nistagmo en el punto final. Los nervios ópticos parecían sanos, con márgenes distintos y una relación copa / disco de 0,15 en ambos ojos.
Dos días después, el paciente declaró una mejoría en todos los síntomas y que sus ojos «se sienten mejor hoy de lo que han estado en un tiempo.»El dolor ocular y la diplopía binocular se resolvieron. Las acuidades visuales permanecieron 20/20 en los ojos derecho e izquierdo. El tamaño de las pupilas fue de 7,5 mm en la oscuridad y de 7,0 mm en la luz, con mejores respuestas directas y consensuadas en ambos ojos. Las motilidades extraoculares estaban llenas en todos los cuadrantes sin dolor ocular. Las búsquedas horizontales siguieron siendo sacádicas con movimientos irregulares y nistagmo en el punto final. El examen neurológico reveló reflejos anormales, que permanecieron estables. Las opciones de tratamiento para la MFS incluyen plasmaféresis e inmunoglobulina IgG intravenosa (IV), pero se retuvieron debido a la mejoría de la oftalmoplejía y la ataxia.
Cuatro días después, el paciente informó que todos los síntomas seguían mejorando, con alguna dificultad para enfocar y rastrear mientras usaba la computadora. El equilibrio se resolvió en un 80% y hubo pocas apariciones de diplopía a distancia. Las acuidades permanecieron 20/20 OU. Las pupilas fueron de 5,5 mm en la oscuridad y de 4,0 mm en la luz con más de 3 respuestas directas y consensuadas UD. Las motilidades extraoculares estaban llenas en todos los cuadrantes sin dolor ocular. Se mejoraron las búsquedas horizontales, sin movimientos sacádicos. La acomodación monocular fue normal, que se midió 8,5 y 9 dioptrías prismáticas DO y SG, respectivamente.
Una semana después, el paciente reportó que su visión volvió a ser casi normal sin diplopía y dificultades leves de enfoque y seguimiento. El saldo se resolvió al 100%. Las acuidades visuales permanecieron 20/20 en los ojos derecho e izquierdo. El tamaño de las pupilas fue de 5,5 mm en la oscuridad, 4,0 mm en la luz con más de 4 respuestas directas y consensuadas UD. Las motilidades extraoculares estaban llenas en todos los cuadrantes sin dolor ocular. Las búsquedas horizontales mejoraron sin movimientos sacádicos observados. La salud ocular era normal.
El tratamiento se suspendió debido a la resolución de la oftalmoplejía y ataxia con areflexia estable. La recuperación completa de la oftalmoplejía y la ataxia llevó dos semanas. El tratamiento incluyó una estrecha vigilancia y seguimiento en cuatro semanas; se aconsejó al paciente que regresara antes si reaparecía alguno de sus síntomas.
Características clínicas
En estudios bien descritos de MFS, todos los pacientes presentaron la tríada clínica de oftalmoplejía, ataxia y areflexia.4,5 El inicio de los síntomas suele ocurrir durante varios días, y los pacientes sufren una infección viral de una a cuatro semanas antes del inicio de los síntomas clínicos.4,5 Según un estudio de 50 pacientes con la enfermedad, el intervalo medio entre el inicio de la infección y el desarrollo de los síntomas neurológicos es de ocho días.4 Otros signos de síndrome de fatiga crónica incluyen dificultad para hablar, dificultad para tragar y expresión facial anormal con incapacidad para sonreír o silbar.
Características oftalmológicas. Los médicos deben estar atentos a la midriasis, retracción del párpado, cierre de ángulo agudo y diplopía secundaria a parálisis nerviosa que afectan a CN III, IV y VI3, 8, 10, 22, 24.La diplopía es el síntoma oftalmológico más común reportado en muchos estudios de MFS.8,16,25,26,27 La diplopía también es el síntoma inicial notificado en 38,6% de 223 pacientes en un estudio histórico y 65% de 466 pacientes en un segundo estudio.8,27 En el segundo estudio, el 100% de los pacientes presentaron oftalmoplejía externa, mientras que la oftalmoplejía interna estaba presente en el 35% de los pacientes.27 La oftalmoplejía externa se refiere al deterioro de los músculos extraoculares externos. La oftalmoplejía interna se refiere al deterioro del esfínter pupilar y del músculo ciliar. La oftalmoplejía completa afecta a los músculos externos e internos.
En un estudio retrospectivo de 19 pacientes con MFS, todos presentaron una o más parálisis nerviosa que afectaban los músculos extraoculares.25 Otros estudios muestran que los pacientes con MFS pueden experimentar múltiples parálisis de los nervios craneales, que pueden manifestarse bilateralmente.22,24,25 Un hallazgo común encontrado entre los trastornos positivos para IgG anti-GQ1b es el déficit de abducción consistente con una parálisis CN VI unilateral o bilateral.26,28 La oftalmoplejía interna causa midriasis pupilar, donde la constricción de la pupila a la luz y / o la estimulación cercana puede variar de mínima a ausente.8,26 En un informe, la midriasis estaba presente en el 42% de los pacientes. La afectación de la CN VII ocurrió en aproximadamente el 45,7% de los pacientes con MFS, causando debilidad facial, incapacidad para sonreír o silbar.8
Ataxia. La causa de esta característica clínica en la MFS no se conoce completamente. Existe un debate sobre si la ataxia es causada por una disfunción central en el cerebelo o periférica. La primera hipótesis, propuesta por el Dr. Fisher, postulaba la implicación de neuronas Ia aferentes.2 Esto fue apoyado por un segundo estudio décadas más tarde, que mostró anormalidades de fibras aferentes de Ia en MFS.11 También se ha propuesto una afectación anormal de los nervios periféricos con un desajuste entre el aporte propioceptivo de los husos musculares y la información cinestésica de los receptores articulares.12
También ha habido evidencia de que la ataxia se origina en el cerebelo. Se encontró un predominio de anticuerpos anti-GQ1b IgG en el cerebelo mediante tinción inmunocitoquímica del cerebelo humano y se confirmó con estudios de western blot, que mostraron un aumento de anticuerpos anti-GQ1b IgG en pacientes con MFS en comparación con el control, aunque no se ha estudiado el mecanismo de compromiso cerebeloso.13,14 En 2015, los investigadores publicaron un informe de caso de un paciente con MFS que se sometió a una resonancia magnética. Hubo una relación reducida de N-acetilaspartato / creatina, lo que sugiere disfunción cerebelosa, que volvió a la normalidad 2,5 meses después de la recuperación.23
Areflexia. Esta característica clínica, junto con la depresión de los reflejos tendinosos profundos, es un signo de compromiso del sistema nervioso periférico en la MFS. Los estudios electrofisiológicos han validado la conducción nerviosa periférica anormal en SGB y SME.3 En un estudio, el 81,6% de los casos de MFS presentaron arefexia.8 Otro estudio de referencia mostró que todos los pacientes experimentaron areflexia, que todavía estaba presente seis meses después de su aparición.4
¿Qué tan común es el MFS?
Los datos epidemiológicos sobre el síndrome de Miller Fisher son limitados. La incidencia de MFS es bastante rara, y ocurre en 0,09 por cada 100.000 personas al año.3 La MFS se presenta con más frecuencia en pacientes con síndrome de Guillain-Barré, del 3% al 25%.4,6,7 Esta asociación ocurre más en pacientes de ascendencia del Lejano Oriente, lo que sugiere un posible componente genético de la MFS. La edad media de inicio es de 43 años, con un rango de 13 a 78,5 MFS que afecta a los hombres dos veces más que a las mujeres, con una proporción de dos a 1,03,5 MFS que ocurre más en las temporadas de invierno y primavera.5,6,8 No se ha determinado la causa exacta de la predilección estacional, pero es probable que esté asociada con la naturaleza postinfecciosa de la afección, ya que se ha encontrado que las infecciones bacterianas y virales desencadenan una respuesta autoinmune que causa la producción del anticuerpo IgG anti-GQ1b en la MFS.
Fisiopatología
La fisiopatología del síndrome de Miller Fisher no se conoce bien; sin embargo, existen varias hipótesis a partir de estudios inmunológicos e histológicos. Es bien sabido que la afección se encuentra dentro del espectro de síndromes de anticuerpos IgG anti-GQ1b junto con el síndrome de Guillain-Barré y la encefalitis del tronco encefálico de Bickerstaff.
El gangliósido GQ1b es un grupo de lípidos complejos que participan en el sistema nervioso central y periférico. GQ1b es un componente de la estructura de la membrana plasmática en los nervios craneales que abastecen los músculos extraoculares y está involucrado en la función celular en la unión neuromuscular presináptica.
Un mecanismo autoinmune de una infección desencadenante anterior producirá el anticuerpo IgG anti-GQ1b, que daña la función gangliósida GQ1b, causando desmielinización. Los estudios histológicos han mostrado desmielinización e hinchazón axonal en los nervios craneales periféricos y oculomotores.15 El anticuerpo anti-GQ1bIgG está ausente en pacientes sanos, pero positivo en más del 90% de los pacientes con MFS.16,25
Se ha encontrado que las infecciones bacterianas y virales desencadenan una respuesta autoinmune, causando la producción del anticuerpo IgG anti-GQ 1b. Los siguientes agentes infecciosos que se han asociado con MFS incluyen neumonía por micoplasma, VIH, Campylobacter jejuni, gripe hemofílica, Helicobacter pylori y virus de Epstein-Barr.4,16 En un informe que analizó a 466 pacientes con MFS, el 90% tenía una enfermedad antecedente, que incluía infección de las vías respiratorias superiores, diarrea o ambas.27
Pronóstico y tratamiento
El síndrome de Miller Fisher es una enfermedad autolimitada y tiene un pronóstico general positivo. Los síntomas generalmente mejoran después de unas pocas semanas, con una recuperación completa que suele ocurrir en dos o tres meses. En un estudio, la recuperación comenzó en una mediana de 13 días desde el inicio de los síntomas, y la resolución completa de la oftalmoplejía y la ataxia se produjo en seis meses.Se han encontrado 4,5 recaídas en menos del 3% de los casos.4,5
Aunque la MFS es típicamente autolimitante, las complicaciones sistémicas se han asociado con la afección. Se han encontrado infecciones de las vías respiratorias superiores en 56 a 76% de los pacientes con MFS, que pueden progresar a insuficiencia respiratoria que requiere ventilación mecánica.4,18,19 Otras complicaciones graves poco frecuentes incluyen miocardiopatía, acidosis láctica y coma.3,20
Las opciones de tratamiento para la MFS incluyen plasmaféresis, inmunoglobulina IgG intravenosa y monitoreo sin tratamiento. Los investigadores postulan que la plasmaféresis y la IgG IV pueden ser efectivas para acelerar el tiempo de resolución, ya que el anticuerpo Gq1b es de clase IgG y la semivida de IgG es de aproximadamente 21 días, que es más larga que las semividas de cinco a seis días de IgM e IgA.19 Sin embargo, no se han realizado estudios aleatorizados doble ciego controlados con placebo que investiguen estos tratamientos.
Un estudio retrospectivo no demostró que la plasmaféresis cambiara la probabilidad de recuperación completa y el tiempo para resolver la ataxia y la oftalmoplejía; además, el intervalo entre el inicio y el inicio del tratamiento con plasmaféresis no afectó el tiempo hasta la resolución ni la gravedad de los síntomas al inicio. Sin embargo, un reporte de caso indica que la plasmaféresis está indicada cuando hay ataxia profunda, insuficiencia motora y respiratoria.21
Los pacientes deben ser monitorizados estrechamente para detectar el riesgo de desarrollar complicaciones sistémicas graves, como insuficiencia respiratoria superior. Los estudios no muestran una mejora significativa en el tiempo de recuperación con el tratamiento de IgG IV o plasmaféresis en MFS, por lo que el monitoreo puede ser la mejor opción para un profesional.
Aunque es poco frecuente, la afección no necesita ser diagnosticada. Al reconocer las características clínicas clave, los profesionales pueden revelar la causa de los síntomas de su paciente y ganar confianza en una recuperación completa.
Los doctores Wang y Cantrell son optometristas del personal del Orlando VA Medical Center. El Dr. Cali es optometrista del personal de la Clínica VA del Condado de Lee. Los autores desean agradecer a Joseph Miller, OD, Paul Gruosso, OD, y Vanessa Santos, OD, por su ayuda con este manuscrito.
1. Collier J. Neuritis periférica: The Morisa lectures (en inglés). Edinburgh Med J. 1932: 39: 601-18.
2. Fisher M. Una variante inusual de la polineuritis idiopática aguda (síndrome de oftalmoplejía, ataxia y areflexia). N Eng J Med. 1956:255:57-65.
3. Lo YL, et al. Espectro clínico e inmunológico del síndrome de Miller Fisher. Nervio Muscular. 2007:36(5):615-27.
4. Mori M, Kuwabara S, Fukutake T, et al. Características clínicas y pronóstico del Síndrome de Miller Fisher. Neurología. 2001:56(8): 1105-6.
5. Mori M, Kuwabara S, Fukutake T, et al. Plasmafaresia y síndrome de Miller Fisher: Análisis de 50 casos consecutivos. Psiquiatría de Neurocirugía Neurológica. 2002:72:680.
6. Yuan CL, Wang YJ, Tsai CP. Síndrome de Miller Fisher: Un estudio retrospectivo basado en hospitales. Euro J Neurol. 2000:44(2):79-85.
7. Cheng BC, Chang WN, Chang CS et al. Síndrome de Guillain-Barré en el sur de Taiwán: Características clínicas, factores pronósticos y resultados terapéuticos. Euro J Neurol. 2003:10 (6):655-62.
8. Berlit P, Rakicky J. El síndrome de Miller Fisher. Revisión de la literatura. J neuroftalmología clínica. 1992:12:57-63.
9. Lyu RK, Tang LM, Cheng SY, et al. Síndrome de Guillain-Barré en Taiwán: Un estudio clínico de 167 pacientes. Psiquiatría de Neurocirugía Neurológica. 1997:63:494-500.
10. Tatsomoto M, Odaka M, Hirata K, Yuki N. Parálisis abducente aislada como variante regional del Síndrome de Guillain-Barré. J Neurol Sci. 2006:243:35-8.
11. Weiss JA, White JC. Correlación de la conducción aferente 1a con ataxia del síndrome de Fisher. Nervio Muscular. 1986:9:327-32.12. Das P, Biswash S, et al. Variante Miller Fisher del síndrome de guillain-barré: Reporte de un caso y revisión clínica. Bangabandhu Sheikh Mujib Medical Univer J. 2012: 5(1): 69-71 13. Kornberg AJ, Pestronk A, Blume GM, et al. Tinción selectiva de la capa molecular cerebelosa por IgG sérica de Miller Fisher y síndromes relacionados. Neurología. 1996:47:1317-20.14. Inoue N, Koh C, Iwahashi T. Detección de anticuerpos anticerebelosos séricos en pacientes con síndrome de Miller Fisher. Euro J Neurol. 1999:42:230-4.15. Barontini F, Di Lollo S, Maurri S, Lambruschini P. Localización del proceso patológico en el Síndrome de Miller Fisher. Italia J Neurol Sci. 1992:13:221-5.16. Koga M, Yuki N, Takahashi M, et al. Estrecha asociación de anticuerpos antigangliósidos IgA con antecedentes de infección por Campylobacter jejuni en síndromes de Guillain-Barré y Fisher. J Neuroinmunol. 2003:141:112-7.17. Guiloff RJ. Conducción nerviosa periférica en el síndrome de Miller Fisher. J Neurol Neurosurg Psychiat. 1977:40:801-7.18. Chikakiya H, Kunishige M, Yoshino H, et al. Neuropatía motora y sensorial retardada en un paciente con encefalitis del tronco encefálico. J Neurol Sci. 2005:234:105-8.19. Arakata Y, Yoshimuro M, Kobayashi S, et al. El uso de inmunoglobulina intravenosa en el síndrome de Miller Fisher. Desarrollo Cerebral J. 1993: 15:231-3.20. Matsumoto H, Kobayashi O, Tamura K, et al. Síndrome de Miller Fisher con coma transitorio: comparación con la encefalitis del tronco encefálico de bickerstaff. Brain Develop J. 2002: 24: 98-101.21. Yeh JH, Chen WH, Chen JR, et al. Síndrome de Miller Fisher con afectación central: tratamiento exitoso con plasmaféresis. Therap Aféresis Dialysis J. 1999: 3: 69-71.22. Papanikolaou T, Cath G, Boothman B, et al. Reporte de caso: oftalmoparesia bilateral aguda con midriasis de areflexia pupilar en síndrome de Miller-Fisher tratada con inmunoglobulina intravenosa. J Oftalmol. 2010; Número de artículo 291840. 23. Síndrome de Sandler RD, Hoggard N, Hadjivassiliou M. Miller-Fisher: ¿La ataxia es central o periférica? Ataxias Cerebelosas. 2015:2:3.24. Ejma M, Waliszewska-Prosół M, Hofman A, et al. Síndrome de Miller-fisher subagudo progresivo tratado con éxito con plasmaféresis. Neurocirugía Neurológica Polaca. 2015;49(2):137-8.25. San-Juan OD, Martínez-Herrera JF, Moreno García J, et al. Síndrome de Miller Fisher: 10 años de experiencia en un centro de tercer nivel. Euro Neurol. 2009; 62: 149-54 26. Snyder LA, Rismondo V, Miller NR. La variante Fisher del síndrome de Guillain-Barré (síndrome de Fisher). J Neurooftalmol. 2009 Dec; 29 (4): 312-24.27. Ku M, Kuwabara S, Odaka M, et al. La encefalitis del tronco encefálico de Bickerstaff y el síndrome de Fisher forman un espectro continuo: análisis clínico de 581 casos. J Neurol. 2008;255:674-82.28. Yuki N. Paresia aguda de los músculos extraoculares asociada con anticuerpos IgG anti-GQ1b. Ann Neurol 1996; 39: 668-72.