Schwach in den Knien: Miller-Fisher-Syndrom
Das Miller-Fisher-Syndrom (MFS), eine seltene, selbstlimitierende Variante des Guillain-Barré-Syndroms (GBS), ist ein Anti-GQ1bIgG-Antikörpersyndrom, das das periphere und zentrale Nervensystem betrifft.1 Als akuter neuromuskulärer polyneuropathischer Zustand verursacht MFS eine aufsteigende Lähmung mit einer klassischen Präsentationstrias aus Ophthalmoplegie, Ataxie und Areflexie. Forscher beschrieben die klinische Triade erstmals 1932.1 Zwei Jahrzehnte später veröffentlichte der kanadische Schlaganfallspezialist Charles Miller Fisher 1956 als erster einen Bericht, in dem die klinischen Befunde beschrieben und der Zustand als begrenzte Form von GBS definiert wurde.2
Symptome treten über mehrere Tage auf, oft im Winter und Frühling, typischerweise nach einer Virusinfektion, und Patienten erleben häufig Diplopie als eines der ersten Symptome. Wenn ein Patient Symptome wie intermittierende Esotropie, beeinträchtigte horizontale Aktivitäten und feste mydriatische Pupillen aufweist und Ihre neurologische Untersuchung abnormal ist, ist es an der Zeit, an GBS, insbesondere MFS, zu denken. Der folgende Fall veranschaulicht die relevanten Anzeichen und Symptome und spiegelt das typische Ergebnis wider.
Geschichte
Ein 27-jähriger kaukasischer Mann, der in den Frühlingsmonaten der VA Eye Clinic vorgestellt wurde, berichtete über eine akute binokulare Diplopie, die einen Tag zuvor begonnen hatte. Der Patient gab an, dass es konstant war, aber nur bei Distanzbetrachtung mit horizontal verschobenen Objekten auftrat. Es gab keine Augenschmerzen oder verschwommenes Sehen im Zusammenhang mit der Diplopie. Er klagte auch über Schwindel und Gleichgewichtsstörungen. Er berichtete von einer Nasennebenhöhlenentzündung mit signifikanter Nasennebenhöhlenverstopfung, die eine Woche vor dem Doppelsehen begann. Frühere Augen- und Krankengeschichten waren unauffällig; Der Patient nahm derzeit keine Medikamente ein und hatte keine Medikamentenallergien.
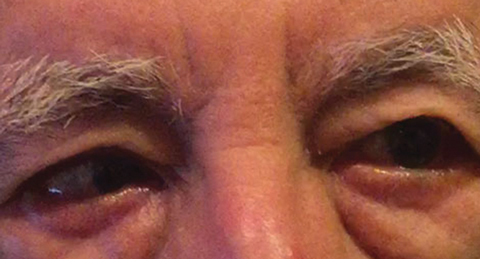
Abb. 1. Die Differentialdiagnose nach der Baseline-Untersuchung umfasste eine partielle rechte CN-VI-Lähmung, die auf einen vaskulären Zustand hinweisen könnte, wie hier bei einem anderen Patienten. Klicken Sie auf das Bild, um es zu vergrößern. Foto: Michael DelGiodice, OD
Diagnosedaten
Bei der Baseline-Untersuchung betrug die unkorrigierte Sehschärfe 20/20-2 OD und 20/25-2 OS. Die Pupillen waren 5 mm gleich rund und reagierten auf Licht ohne afferenten Pupillendefekt. Die extraokularen Motilitäten waren in beiden Augen ohne Einschränkungen voll, glatt und genau. Ein Entfernungstest ergab eine intermittierende Vier- bis Sechs-Prisma-Dioptrien-Rechts-Esotropie im primären Blick, die ungefähr 50% der Zeit auftrat. Die Durchführung eines Tests in der Nähe ergab eine Vier-Prisma-Dioptrien-Astoria im primären Blick ohne Tropie.
Obwohl in diesem Fall nicht durchgeführt, sollten Kliniker den Test im primären, rechten und linken Blick abdecken. Bei Patienten mit Diplopie manifestiert sich die Lähmung des Hirnnervensystems (CN VI) als nicht komitante Abweichung, schlimmer im rechten oder linken Blick, je nachdem, auf welcher Seite sich die Lähmung befindet. Eine dekompensierende Phorie zeigt dagegen eine komitante Abweichung.
Der Brechungsfehler betrug -0,50D sphere OD und -0,75D sphere OS und das Sehvermögen war in jedem Auge auf 20/20 korrigierbar. Der Augeninnendruck lag in beiden Augen innerhalb normaler Grenzen. Die Befunde des vorderen und hinteren Segments waren im rechten und linken Auge unauffällig. Die Sehnerven erschienen gesund mit einem Cup-to-Disc-Verhältnis von 0,15 in beiden Augen.
Differentialdiagnosen umfassten eine partielle rechte CNI-Lähmung (Abbildung 1), Myasthenia gravis, einen kompressiven Prozess und eine dekompensierende rechte Ösophorie, die eine intermittierende rechte Esotropie verursachte. Der Patient bestritt jede Vorgeschichte einer binokularen Diplopie.Die Bildgebung des Gehirns und der Bahnen wurde von der Augenklinik angeordnet, um eine Beteiligung des Gehirns, einschließlich einer kompressiven Läsion, auszuschließen. Computertomographie (CT) -Scans des Gehirns und der Umlaufbahnen wurden vom Neuroradiologen des Personals durchgeführt und interpretiert. CT wurde aufgrund des Timings und der Verfügbarkeit anstelle der Magnetresonanztomographie (MRT) gewählt; Tests und Ergebnisse konnten am selben Tag mit CT in der Klinik nach Stunden erhalten werden. Die Ergebnisse waren unauffällig ohne Anzeichen einer intrakraniellen Blutung, einer kompressiven Läsion oder eines akuten Infarkts.
Die CT der Nasennebenhöhlen ergab eine mittelschwere bis schwere chronische Nasennebenhöhlenerkrankung und eine vollständige Trübung der rechten Kieferhöhle. Es gab keine Verlängerung in die Umlaufbahn. Der Hausarzt des Patienten wurde über die Symptome, die Befunde der Augenuntersuchung und die CT-Bildgebungsergebnisse des Patienten informiert und begann mit der Behandlung der Sinusitis maxillaris mit einer oralen Azithromycin-fünftägigen Dosispackung und wies den Patienten an, die Augenklinik zu konsultieren.
Der Patient kehrte fünf Tage später nach Abschluss seines oralen Azithromycin-Kurses zurück und gab an, dass er keine Verbesserung der Ferndiplopie erlebte; Darüber hinaus erlebte er das Auftreten von signifikanter Photophobie und periokularen Schmerzen um beide Augen. Der Patient gab an, dass sich auch sein Gangungleichgewicht und seine Gehschwierigkeiten verschlechterten. Bei der Untersuchung blieben die am besten korrigierten Sehschärfen im rechten und linken Auge 20/20. Die Pupillen wurden um 8,5 mm erweitert, ohne dass in beiden Augen Lichtreaktionen auftraten (Abbildung 2). Extraokulare Motilitäten zeigten eine laterale Einschränkung mit Schmerzen bei Bewegung im rechten Auge und eine mediale Einschränkung mit Schmerzen bei Bewegung im linken Auge im Einklang mit einer Lähmung des rechten Blicks (Abbildung 3). Horizontale und vertikale Verfolgungen waren sakkadisch mit ungenauen und ruckartigen Bewegungen. Anteriore und posteriore Segmentbefunde blieben unauffällig OU.
Differentialdiagnosen nach ophthalmologischer Untersuchung umfassten linke internukleäre Ophthalmoplegie, rechte Blickparese und rechte Blickparese mit linker internukleärer Ophthalmoplegie, was für extraokulare Motilitätsbefunde verantwortlich sein könnte. Andere Differentiale sind eine Mittelhirnläsion oder Myasthenia gravis. Differentialdiagnosen für die Pupillenerweiterung umfassen Lähmung des dritten Nervs (CN III), Trauma und pharmakologische Dilatation. Der Patient hatte keine anderen Befunde, die mit einer dritten Nervenlähmung übereinstimmten, und leugnete ein Trauma oder eine Exposition gegenüber pharmakologischen Wirkstoffen.
Der Patient wurde zur weiteren Beurteilung an die Neurologie überwiesen. Die neurologische Untersuchung ergab, dass der Patient vollständig wachsam und orientiert war, ohne das Gesichtsgefühl zu verlieren. Der Patient zeigte eine leichte Ataxie mit Schwierigkeiten, im Tandem zu gehen und aus einer hockenden Position zu stehen. Proximale Schwäche sowohl der oberen als auch der unteren Extremitäten und abnormale Reflexe waren vorhanden. Alle sensorischen Modalitäten waren intakt. MRT und Magnetresonanzangiographie (MRA) des Gehirns und der Bahnen waren unauffällig. Cerebrospinalflüssigkeitsprotein wurde bei 75 mg / dl erhöht (Referenzbereich: 15 mg / dl bis 45 mg / dl) ohne andere Anomalien. Elektromyogramm-Tests waren unauffällig und zeigten keine Anomalien in der Nervenleitung von der Wirbelsäule zu den Füßen und Händen.
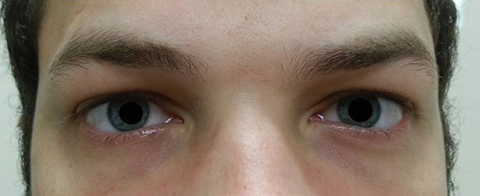
Abb. 2. Die Pupillen des Patienten waren um 8,5 mm erweitert, ohne dass das rechte oder linke Auge reaktiv war. Klicken Sie auf das Bild, um es zu vergrößern.
Diagnose
Der Patient zeigte akute Symptome von Diplopie, Ataxie und Areflexie. Angesichts der intermittierenden rechten Esotropie, beeinträchtigte horizontale Verfolgungen, feste mydriatische Pupille, leichte Ataxie und abnormale Reflexe bei neurologischer Untersuchung und erhöhtes Liquorprotein, Die Diagnose MFS wurde gestellt. Der Patient wurde ins Krankenhaus eingeliefert, um weitere Komplikationen zu überwachen. Eine Augenklappe wurde dem Patienten zur Verfügung gestellt, um das rechte Auge zu bedecken, wie es für Diplopie benötigt wird.
Derzeit gibt es keine veröffentlichten diagnostischen Kriterien für das Miller-Fisher-Syndrom. Die Diagnose wird in der Regel durch die Präsentation der klinischen Triade zusammen mit Bildgebungs- und Liquoruntersuchungen gestellt. MRT ist in der Regel unauffällig in der Bedingung, obwohl einige Studien zentrale Läsionen im Mittelhirn, Pons und unteren Medulla gezeigt haben. Elektrophysiologische Studien zeigen eine verminderte oder abnormale periphere sensorische Überleitung.17
Das Protein der Zerebrospinalflüssigkeit ist häufig erhöht, ohne dass andere abnormale Befunde vorliegen. In einer bahnbrechenden Studie zeigten 64,4% der MFS-Patienten ein erhöhtes Protein in der Rückenmarksflüssigkeit.8 Ein positiver Anti-GQ1b-IgG-Antikörpertest ermöglicht eine definitive Diagnose. In einer Studie war der Anti-GQ1b-IgG-Antikörper bei bis zu 95% der Patienten mit der Erkrankung vorhanden und bei den Kontrollen nicht vorhanden.3
Der Neurologe diagnostizierte den Patienten mit MFS basierend auf der klinischen Trias von Ophthalmoplegie, Ataxie und Areflexie, einer unauffälligen MRT und MRA des Gehirns und erhöhtem Liquorprotein. Daher wurde in diesem Fall kein Anti-GQ1b-IgG-Antikörpertest durchgeführt.Mehrere klinische Merkmale des MFS werden auch beim Guillain-Barré-Syndrom und der Bickerstaff-Hirnstammenzephalitis beobachtet, was die Diagnose schwierig macht. Patienten mit nur Guillain-Barré präsentieren sich mit Gliedmaßenschwäche, sensorischem Verlust, kranialer Neuropathie und Areflexie ohne ophthalmologische Manifestationen. Patienten mit Bickerstaffs Hirnstammenzephalitis weisen typischerweise Ophthalmoplegie, Ataxie, Hyperreflexie und Bewusstseinsstörungen auf. Da bei allen drei Erkrankungen mehrere Symptome und Anzeichen vorliegen, ist der Anti-GQ1b-IgG-Antikörpertitertest für eine definitivere Diagnose hilfreich. Der Anti-GQ1b-IgG-Antikörper ist bei einer großen Mehrheit der Patienten mit Miller-Fisher-Syndrom positiv, verglichen mit 66% bei Bickerstaff-Hirnstammenzephalitis und 26% bei Patienten mit Guillain-Barré-Syndrom.16
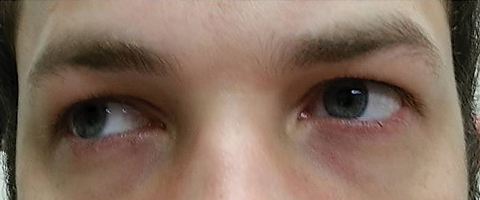
Abb. 3. Extraokulare Motilitäten zeigten eine Einschränkung der Dextroversion. Klicken Sie auf das Bild, um es zu vergrößern.
Behandlung und Nachsorge
Der Patient wurde am Tag nach der Aufnahme untersucht, als er angab, keine visuellen Veränderungen oder eine Verbesserung des Gleichgewichts zu erfahren. Die binokulare Diplopie war immer noch konstant, daher blieb das rechte Auge geflickt. Augenschmerzen mit weiten Blicken und schnellen Augenbewegungen waren immer noch vorhanden. Bei begrenzter Untersuchung am Krankenbett blieben die am besten korrigierten Sehschärfen im rechten und linken Auge 20/20. Die Pupillen wurden um 8 erweitert.0mm in beiden Augen mit minimaler Reaktivität, die gegenüber dem Vortag leicht verbessert wurde. Das Farbsehen war im rechten und linken Auge normal. Extraokulare Motilitäten waren in allen Quadranten voll mit Schmerzen, die bei weiten horizontalen Blicken noch vorhanden waren. Horizontale Verfolgungen blieben sakkadisch mit ruckartigen Bewegungen und Endpunkt-Nystagmus. Die Sehnerven erschienen gesund mit deutlichen Rändern und einem Verhältnis von Tasse zu Scheibe von 0,15 in beiden Augen.
Zwei Tage später gab der Patient eine Besserung aller Symptome an und dass sich seine Augen „heute besser fühlen als seit einiger Zeit.“ Augenschmerzen und binokulare Diplopie wurden beide gelöst. Die Sehschärfe blieb im rechten und linken Auge 20/20. Die Pupillengrößen betrugen 7,5 mm bei Dunkelheit und 7,0 mm bei Licht mit verbesserten direkten und einvernehmlichen Reaktionen in beiden Augen. Extraokulare Motilitäten waren in allen Quadranten ohne Augenschmerzen voll. Horizontale Verfolgungen blieben sakkadisch mit ruckartigen Bewegungen und Endpunkt-Nystagmus. Neurologische Untersuchung ergab abnorme Reflexe, die stabil blieben. Die Behandlungsoptionen für MFS umfassen Plasmapherese und intravenöses (IV) Immunglobulin IgG, aber diese wurden angesichts der Verbesserung der Ophthalmoplegie und Ataxie zurückgehalten.Vier Tage später berichtete der Patient, dass sich alle Symptome noch besserten, mit einigen Schwierigkeiten, sich zu konzentrieren und zu verfolgen, während er den Computer benutzte. Das Gleichgewicht war zu 80% gelöst und es gab nur wenige Fälle von Entfernungsdiplopie. Acuities blieb 20/20 OU. Die Pupillen waren 5,5 mm im Dunkeln und 4,0 mm im Licht mit 3+ direkten und einvernehmlichen Antworten OU. Extraokulare Motilitäten waren in allen Quadranten ohne Augenschmerzen voll. Horizontale Verfolgungen wurden verbessert, ohne sakkadische Bewegungen. Monokulare Akkommodation war normal, die gemessen wurde 8,5 und 9 Prisma Dioptrien OD und OS, beziehungsweise.Eine Woche später berichtete der Patient, dass sein Sehvermögen nahezu normal war, ohne Diplopie und leichte Schwierigkeiten beim Fokussieren und Verfolgen. Balance war 100% gelöst. Die Sehschärfe blieb im rechten und linken Auge 20/20. Pupillengrößen waren 5,5 mm im Dunkeln, 4,0 mm im Licht mit 4+ direkten und einvernehmlichen Antworten OU. Extraokulare Motilitäten waren in allen Quadranten ohne Augenschmerzen voll. Horizontale Verfolgungen verbesserten sich ohne beobachtete sakkadische Bewegungen. Augengesundheit war unauffällig OU.
Die Behandlung wurde aufgrund der Auflösung von Ophthalmoplegie und Ataxie mit stabiler Areflexie zurückgehalten. Die vollständige Genesung von Opthalmoplegie und Ataxie dauerte zwei Wochen. Das Management umfasste eine enge Überwachung und Follow-up in vier Wochen; Dem Patienten wurde geraten, früher zurückzukehren, wenn eines seiner Symptome zurückkehrte.
Klinische Merkmale
In gut beschriebenen Studien zum MFS zeigten alle Patienten die klinische Trias Ophthalmoplegie, Ataxie und Areflexie.4,5 Das Auftreten von Symptomen tritt typischerweise über mehrere Tage auf, und Patienten erleiden eine Virusinfektion ein bis vier Wochen vor dem Auftreten klinischer Symptome.4,5 Laut einer Studie mit 50 Patienten mit dieser Erkrankung beträgt das mittlere Intervall zwischen Infektionsbeginn und Entwicklung neurologischer Symptome acht Tage.4 Andere Anzeichen von MFS sind verschwommene Sprache, Schluckbeschwerden und ein abnormaler Gesichtsausdruck mit der Unfähigkeit zu lächeln oder zu pfeifen.
Ophthalmologische Merkmale. Praktiker sollten auf Mydriasis, Lidretraktion, Spitzwinkelverschluss und Diplopie als Folge von Nervenlähmungen bei CN III, IV und VI achten.3,8,10,22,24 Diplopie ist das häufigste ophthalmologische Symptom, über das in vielen MFS-Studien berichtet wurde.8,16,25,26,27 Diplopie ist auch das erste Symptom, das bei 38,6% von 223 Patienten in einer wegweisenden Studie und 65% von 466 Patienten in einer zweiten Studie berichtet wurde.8,27 In der zweiten Studie zeigten 100% der Patienten eine externe Ophthalmoplegie, während bei 35% der Patienten eine interne Ophthalmoplegie auftrat.27 Externe Ophthalmoplegie bezieht sich auf eine Beeinträchtigung der externen extraokularen Muskeln. Interne Ophthalmoplegie bezieht sich auf eine Beeinträchtigung des Pupillensphinkters und des Ziliarmuskels. Komplette Ophthalmoplegie betrifft sowohl äußere als auch innere Muskeln.
In einer retrospektiven Studie mit 19 Patienten mit MFS, die alle mit einer oder mehreren Nervenlähmungen der extraokularen Muskeln konfrontiert waren.25 Weitere Studien zeigen, dass bei MFS-Patienten multiple Hirnnervenlähmungen auftreten können, die sich bilateral manifestieren können.22,24,25 Ein häufiger Befund bei Anti-GQ1b-IgG-positiven Störungen ist das Abduktionsdefizit im Einklang mit einer einseitigen oder bilateralen CNI-Lähmung.26,28 Interne Ophthalmoplegie verursacht Pupillenmydriasis, bei der die Pupillenverengung durch Licht und / oder nahe Stimulation von minimal bis nicht vorhanden reichen kann.8,26 In einem Bericht war Mydriasis bei 42% der Patienten vorhanden. Die Beteiligung von CN VII trat bei etwa 45,7% der MFS-Patienten auf und verursachte Gesichtsschwäche, Unfähigkeit zu lächeln oder zu pfeifen.8
Ataxie. Die Ursache dieses klinischen Merkmals bei MFS ist nicht vollständig geklärt. Es besteht eine Debatte darüber, ob Ataxie durch Dysfunktion zentral im Kleinhirn oder peripher verursacht wird. Die erste von Dr. Fisher vorgeschlagene Hypothese postulierte die Beteiligung von Ia-afferenten Neuronen.2 Dies wurde Jahrzehnte später durch eine zweite Studie gestützt, die Anomalien der Ia-afferenten Fasern im MFS zeigte.11 Es wurde auch eine abnormale periphere Nervenbeteiligung mit einer Nichtübereinstimmung zwischen propriozeptivem Input von vorgeschlagen Muskelspindeln und kinästhetische Informationen von Gelenkrezeptoren.12
Es gibt auch Belege dafür, dass Ataxie ihren Ursprung im Kleinhirn hat. Ein Vorherrschen von Anti-GQ1b-IgG-Antikörpern im Kleinhirn wurde unter Verwendung einer immunzytochemischen Färbung des menschlichen Kleinhirns festgestellt und weiter mit Western-Blot-Studien bestätigt, die bei MFS-Patienten im Vergleich zur Kontrolle einen erhöhten Anti-GQ1b-IgG-Antikörper zeigten, obwohl der Mechanismus für die Beteiligung des Kleinhirns nicht untersucht wurde.13,14 In 2015 veröffentlichten Forscher einen Fallbericht eines MFS-Patienten, der sich einer MRT unterzog. Es gab ein reduziertes N-Acetylaspartat / Kreatin-Verhältnis, was auf eine zerebelläre Dysfunktion hindeutet, die sich 2,5 Monate nach der Genesung wieder normalisierte.23
Areflexie. Dieses klinische Merkmal ist zusammen mit der Depression tiefer Sehnenreflexe ein Zeichen für eine Beteiligung des peripheren Nervensystems am MFS. Elektrophysiologische Studien haben eine abnormale periphere Nervenleitung bei GBS und MFS bestätigt.3 In einer Studie wiesen 81,6% der MFS-Fälle Arefexie auf.8 Eine weitere wegweisende Studie zeigte, dass bei allen Patienten Areflexie auftrat, die sechs Monate nach Beginn noch vorhanden war.4
Wie häufig ist MFS?
Epidemiologische Daten zum Miller-Fisher-Syndrom sind begrenzt. Die Inzidenz von MFS ist ziemlich selten und tritt bei 0,09 pro 100.000 Menschen pro Jahr auf.3 MFS tritt häufiger bei Patienten mit Guillain-Barré-Syndrom auf, von 3% bis 25%.4,6,7 Diese Assoziation tritt häufiger bei Patienten fernöstlicher Abstammung auf, was auf eine mögliche genetische Komponente des MFS hindeutet. Das mittlere Erkrankungsalter beträgt 43 Jahre, wobei ein Bereich von 13 bis 78,5 MFS Männer doppelt so stark betrifft wie Frauen mit einem Verhältnis von zwei zu 1,03,5 MFS tritt häufiger in den Winter- und Frühlingssaisons auf.5,6,8 Die genaue Ursache der saisonalen Vorliebe wurde nicht bestimmt, ist aber wahrscheinlich mit der postinfektiösen Natur der Erkrankung verbunden, da festgestellt wurde, dass bakterielle und virale Infektionen eine Autoimmunantwort auslösen, die die Produktion des Anti-GQ1b-IgG-Antikörpers im MFS verursacht.
Pathophysiologie
Die Pathophysiologie des Miller-Fisher-Syndroms ist nicht gut verstanden; Es gibt jedoch mehrere Hypothesen aus immunologischen und histologischen Studien. Es ist bekannt, dass die Erkrankung zusammen mit dem Guillain-Barré-Syndrom und der Bickerstaff-Hirnstamm-Enzephalitis im Spektrum der Anti-GQ1b-IgG-Antikörper-Syndrome liegt.
Das Gangliosid GQ1b ist eine Gruppe komplexer Lipide, die am zentralen und peripheren Nervensystem beteiligt sind. GQ1b ist ein Bestandteil der Plasmamembranstruktur in Hirnnerven, die die extraokularen Muskeln versorgen, und ist an der Zellfunktion am präsynaptischen neuromuskulären Übergang beteiligt. Ein Autoimmunmechanismus aus einer vorhergehenden auslösenden Infektion produziert den Anti-GQ1b-IgG-Antikörper, der die Gangliosid-GQ1b-Funktion schädigt und eine Demyelinisierung verursacht. Histologische Studien haben Demyelinisierung und axonale Schwellung in peripheren und okulomotorischen Hirnnerven gezeigt.15 Der Anti-GQ1bIgG-Antikörper fehlt bei gesunden Patienten, ist aber bei über 90% der Patienten mit MFS positiv.16,25
Es wurde festgestellt, dass bakterielle und virale Infektionen eine Autoimmunreaktion auslösen, die die Produktion des Anti-GQ 1b IgG-Antikörpers verursacht. Die folgenden Infektionserreger, die mit MFS in Verbindung gebracht wurden, umfassen Mycoplasma-Pneumonie, HIV, Campylobacter jejuni, Hämophilus Influenza, Helicobacter pylori und Epstein-Barr-Virus.4,16 In einem Bericht, der 466 MFS-Patienten untersuchte, hatten 90% eine Vorerkrankung, einschließlich Infektionen der oberen Atemwege, Durchfall oder beides.27
Prognose und Behandlung
Das Miller-Fisher-Syndrom ist eine selbstlimitierende Erkrankung und hat eine insgesamt positive Prognose. Die Symptome bessern sich im Allgemeinen nach einigen Wochen, wobei die vollständige Genesung typischerweise in zwei bis drei Monaten erfolgt. In einer Studie begann die Erholung im Median 13 Tage nach Beginn der Symptome, und die vollständige Auflösung von Ophthalmoplegie und Ataxie erfolgte in sechs Monaten.Es wurde festgestellt, dass 4,5 in weniger als 3% der Fälle Rückfälle auftraten.4,5
Obwohl das MFS typischerweise selbstlimitierend ist, wurden systemische Komplikationen mit der Erkrankung in Verbindung gebracht. Infektionen der oberen Atemwege wurden bei 56% bis 76% der MFS-Patienten gefunden, die zu Atemversagen führen können, das eine mechanische Beatmung erfordert.4,18,19 Weitere seltene schwerwiegende Komplikationen sind Kardiomyopathie, Laktatazidose und Koma.3,20
Die Behandlungsoptionen für MFS umfassen Plasmapherese, IV-Immunglobulin-IgG und Überwachung ohne Behandlung. Forscher postulieren, dass Plasmapherese und IV IgG wirksam sein können, um die Auflösungszeit zu beschleunigen, da Antikörper Gq1b IgG in der Klasse ist und die Halbwertszeit von IgG ungefähr 21 Tage beträgt, was länger ist als die fünf- bis sechstägigen Halbwertszeiten von IgM und IgA.19 Es wurden jedoch keine randomisierten, doppelblinden, placebokontrollierten Studien zur Untersuchung dieser Behandlungen durchgeführt.Eine retrospektive Studie konnte nicht zeigen, dass die Plasmapherese die Chance auf eine vollständige Genesung und die Zeit bis zum Abklingen von Ataxie und Ophthalmoplegie veränderte; Darüber hinaus hatte das Intervall vom Beginn bis zum Beginn der Plasmapherese-Behandlung keinen Einfluss auf die Zeit bis zum Abklingen oder die Schwere der Symptome zu Beginn. Ein Fallbericht zeigt jedoch, dass eine Plasmapherese indiziert ist, wenn eine tiefgreifende Ataxie, motorische und respiratorische Beeinträchtigung vorliegt.21
Die Patienten sollten engmaschig auf das Risiko der Entwicklung schwerwiegender systemischer Komplikationen wie Versagen der oberen Atemwege überwacht werden. Studien zeigen keine signifikante Verbesserung der Erholungszeit mit IV IgG oder Plasmapherese-Behandlung bei MFS, so dass die Überwachung die beste Option für einen Arzt sein kann.
Obwohl selten, muss der Zustand nicht undiagnostiziert bleiben. Durch das Erkennen der wichtigsten klinischen Merkmale können Ärzte die Ursache der Symptome ihres Patienten aufdecken und Vertrauen in eine vollständige Genesung gewinnen.
Drs. Wang und Cantrell sind Augenoptiker am Orlando VA Medical Center. Dr. Cali ist Augenoptikerin in der Lee County VA Clinic. Die Autoren danken Joseph Miller, OD, Paul Gruosso, OD, und Vanessa Santos, OD, für die Unterstützung bei diesem Manuskript.
1. Collier J. Periphere Neuritis: Die Morisa-Vorlesungen. Edinburgh Med J. 1932:39:601-18.
2. Fisher M. Eine ungewöhnliche Variante der akuten idiopathischen Polyneuritis (Syndrom Ophthalmoplegie, Ataxie und Areflexie). In: N Eng J Med. 1956:255:57-65.
3. In: Lo YL, et al. Klinisches und immunologisches Spektrum des Miller-Fisher-Syndroms. Muskelnerv. 2007:36(5):615-27.
4. Mori M, Kuwabara S, Fukutake T, et al. Klinische Merkmale und Prognose des Miller-Fisher-Syndroms. Neurologie. 2001:56(8): 1105-6.
5. Mori M, Kuwabara S, Fukutake T, et al. Plasmapharese und Miller-Fisher-Syndrom: Analyse von 50 aufeinanderfolgenden Fällen. J Neurol Neurochirurgie Psych. 2002:72:680.
6. Yuan CL, Wang YJ, Tsai CP. Miller-Fisher-Syndrom: Eine retrospektive Studie im Krankenhaus. In: Euro J Neurol. 2000:44(2):79-85.
7. Cheng BC, Chang WN, Chang CS et al. Guillain-Barré-Syndrom in Südtaiwan: Klinische Merkmale, prognostische Faktoren und therapeutische Ergebnisse. In: Euro J Neurol. 2003:10 (6):655-62.
8. Berlit P, Rakicky J. Das Miller-Fisher-Syndrom. Überprüfung der Literatur. J klinische Neuro-Ophthalmologie. 1992:12:57-63.
9. Dr.-Ing. Dr.-Ing. Dr.-Ing. Dr.-Ing.Dr.-Ing. Guillain-Barré-Syndrom in Taiwan: Eine klinische Studie mit 167 Patienten. J Neurol Neurochirurgie Psych. 1997:63:494-500.
10. Tatsomoto M, Odaka M, Hirata K, Yuki N. Isolierte Abducens-Lähmung als regionale Variante des Guillain-Barré-Syndroms. In: J Neurol Sci. 2006:243:35-8.
11. Weiß JA, Weiß JC. Korrelation der 1a-afferenten Überleitung mit der Ataxie des Fisher-Syndroms. Muskelnerv. 1986:9:327-32.
12. Das P, Biswash S, et al. Miller-Fisher-Variante des Guillain-Barré-Syndroms: Ein Fallbericht und eine klinische Überprüfung. Bangabandhu Sheikh Mujib Medizinische Univer J. 2012:5(1):69-71
13. Kornberg AJ, Pestronk A, Blume GM, et al. Selektive Färbung der Kleinhirnmolekülschicht durch Serum-IgG von Miller Fisher und verwandten Syndromen. Neurologie. 1996:47:1317-20.
14. Inoue N, Koh C, Iwahashi T. Nachweis von Serum-Anticerebellar-Antikörpern bei Patienten mit Miller-Fisher-Syndrom. In: Euro J Neurol. 1999:42:230-4.
15. Barontini F, Di Lollo S, Maurri S, Lambruschini P. Lokalisierung des pathologischen Prozesses beim Miller-Fisher-Syndrom. Italien J Neurol Sci. 1992:13:221-5.
16. Koga M, Yuki N, Takahashi M, et al. Enge Assoziation von IgA-Anti-Gangliosid-Antikörpern mit antezedenter Campylobacter-jejuni-Infektion bei Guillain-Barré- und Fisher-Syndromen. In: J Neuroimmunol. 2003:141:112-7.
17. Guiloff RJ. Periphere Nervenleitung beim Miller-Fisher-Syndrom. J Neurol Neurochirurg Psychat. 1977:40:801-7.
18. Chikakiya H., Kunishige M., Yoshino H., et al. Verzögerte motorische und sensorische Neuropathie bei einem Patienten mit Hirnstammenzephalitis. In: J Neurol Sci. 2005:234:105-8.
19. Kobayashi S, Yoshimuro M, Arakata Y, et al. Die Verwendung von intravenösem Immunglobulin beim Miller-Fisher-Syndrom. Gehirn entwickeln J. 1993:15:231-3.
20. Matsumoto H, Kobayashi O, Tamura K, et al. Miller-Fisher-Syndrom mit transientem Koma: Vergleich mit Bickerstaffs Hirnstammenzephalitis. Gehirn entwickeln J. 2002:24:98-101.
21. Yeh JH, Chen WH, Chen JR, et al. Miller-Fisher-Syndrom mit zentraler Beteiligung: erfolgreiche Behandlung mit Plasmapherese. Therap Apherese Dialyse J.1999: 3:69-71.
22. Papanikolaou T, Kath G, Boothman B, et al. Fallbericht: Akute bilaterale Ophthalmoparese mit pupilärer areflexischer Mydriasis beim Miller-Fisher-Syndrom, behandelt mit intravenösem Immunglobulin. J Ophthalmol. 2010;Artikelnummer 291840.
23. Sandler RD, Hoggard N, Hadjivassiliou M. Miller-Fisher-Syndrom: Ist die Ataxie zentral oder peripher? Kleinhirnataxie. 2015:2:3.
24. Ejma M, Waliszewska-Prosół M, Hofman A, et al. Progressive subakute Miller-Fisher-Syndrom erfolgreich mit Plasmapherese behandelt. Polnische J Neurol Neurochirurgie. 2015;49(2):137-8.
25. San-Juan OD, Martínez-Herrera JF, Moreno García J, et al. Miller-Fisher-Syndrom: 10 Jahre Erfahrung in einem Third-Level-Center. In: Euro Neurol. 2009;62:149-54
26. Snyder LA, Rismondo V, Miller NR. Die Fisher-Variante des Guillain-Barré-Syndroms (Fisher-Syndrom). In: J Neuroophthalmol. 2009 Dezember;29(4):312-24.
27. Ito M, Kuwabara S, Odaka M, et al. Bickerstaffs Hirnstammenzephalitis und das Fisher-Syndrom bilden ein kontinuierliches Spektrum: klinische Analyse von 581 Fällen. J Neurol. 2008;255:674-82.
28. Yuki N. Akute Parese der extraokularen Muskeln im Zusammenhang mit IgG-Anti-GQ1b-Antikörper. Ann Neurol 1996;39:668-72.